Chapter: Pathology: Genetic Disorders
Autosomal Dominant Disorders
AUTOSOMAL DOMINANT DISORDERS
Familial
hypercholesterolemia is the most common inherited
disorder (incidence1 in 500) and is due to a mutation in the low density
lipoprotein (LDL) receptor gene (LDLR) on chromosome 19. The mutations in the
LDL receptor cause increased levels of circulating cholesterol, loss of
feedback inhibition of HMG-coenzyme A (HMG-CoA) reductase, and increased
phagocytosis of LDL by macrophages.
There
are 5 major classes of mutation.
·
Class
I: no LDL receptor synthesis
·
Class
II: defect in transport out of the endoplasmic reticulum
·
Class
III: defect in LDL receptor binding
·
Class
IV: defect in ability to internalize bound LDL
·
Class
V: defect in the recycling of the LDL receptor
Clinical
features include elevated serum cholesterol (heterozygotes have elevations of
2–3 times the normal level and homozygotes have elevations of 5–6 times the
normal level), skin xanthomas (collections of lipid-laden macrophages),
xanthelasma around the eyes, and premature atherosclerosis (homozygotes often
develop myocardial infarctions in late teens and twenties).
Marfan
syndrome is due to a mutation of thefibrillingene (FBN1) on
chromosome15q21. Fibrillin is a glycoprotein that functions as a scaffold for
the alignment of elastic fibers.
Clinical
features include skeletal changes (tall, thin build with long extremities,
hyperextensible joints, pectus excavatum [inwardly depressed sternum], and
pec-tus carinatum [pigeon breast]) and abnormal eyes (ectopia lentis,
characterized by bilateral subluxation of the lens). The cardiovascular system
is also particularly vulnerable; it may show cystic medial degeneration of the
media of elastic arteries with a loss of elastic fibers and smooth muscle cells
with increased risk of dissecting aortic aneurysm (a major cause of death),
dilatation of the aortic ring potentially leading to aortic valve
insufficiency, and/or mitral valve prolapse.
Ehlers-Danlos
syndrome (EDS) is a group of inherited connective tissue diseases
thathave in common a defect in collagen structure or synthesis. Clinically, the
disease causes hyperextensible skin that is easily traumatized and
hyperextensible joints sec-ondary to effects on the joints and adjacent
ligaments.
There
are a number of variants with different modes of inheritance.
·
Kyphoscoliotic
EDS: autosomal recessive form
·
Vascular
variant EDS: autosomal dominant form that causes rupture of ves-sels
and bowel wall
·
Classical
EDS: autosomal dominant form that causes a type V collagen
defect;patients have a normal lifespan
Neurofibromatosis
Type
1 (von
Recklinghausen disease) neurofibromatosis (90% of cases)
has an inci-dence of 1 in 3,000. The condition is due to a mutation of the
tumor suppressor gene NF1 located on
chromosome 17 (17q11.2). The normal gene product (neurofibromin)inhibits p21
ras oncoprotein.
·
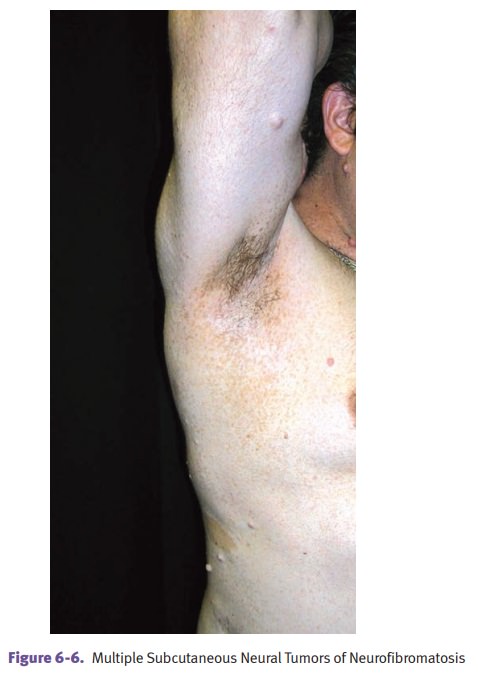
Affected individuals
characteristically have multiple neurofibromas and benign tumors of peripheral
nerves that are often numerous and may be disfiguring. The plexiform variant of
the neurofibromas are diagnostic.
·
Rarely (3%), malignant transformation
of a neurofibroma may occur.
·
Other clinical features include
pigmented skin lesions (6 or more “cafe-au-lait spots” that are light brown
macules usually located over nerves); pigmented iris hamartomas (Lisch
nodules); and increased risk of meningiomas and pheo-chromocytoma, an adrenal
tumor that also occurs with von Hippel-Lindau disease and MEN 2.
Type
2 (bilateral
acoustic) neurofibromatosis (10% of cases) has an incidence of 1 in
45,000.
·
There is a mutated tumor suppressor
gene NF-2 (22q12.2) on chromosome 22.
·
The normal gene product (merlin) is
a critical regulator of contact-dependent inhibition of proliferation.
·
Clinical features include vestibular
schwannomas (acoustic neuromas), and increased risk of meningioma and
ependymomas.
von
Hippel-Lindau disease is due to a mutation of the tumor
suppressor geneVHLon chromosome 3p
(3p26-p25). The normal gene product’s main action is to tag proteins (e.g.,
hypoxia inducible factor 1a [a transcription factor that induces the expression
of angiogenesis factors]) with ubiquitin for degradation.
Clinical manifestations can include retinal hemangioblastoma (von Hippel tumor); hemangioblastomas of the cerebellum, brain stem, and spinal cord (Lindau tumor); cysts of the liver, pancreas, and kidneys; and multiple bilateral renal cell carcinomas.
Related Topics