Chapter: Medical Immunology: Organ-Specific Autoimmune Diseases
Autoimmune Diseases of the Pancreas
AUTOIMMUNE DISEASES OF THE PANCREAS
A. Diabetes Mellitus
Diabetes mellitus (DM) is a multiorgan disease with multiple etiologies and certainly with more than one basic abnormality. In type 1 diabetes (autoimmune diabetes), the basic de-fect is a decreased to absent production of insulin secondary to β-cell destruction (for which reason it is also known as insulin-dependent DM, or IDDM). In type 2 diabetes, there is a decrease in the effect of insulin at the target cell level.
1. Pathogenesis
The major body of evidence on which the concept of type 1 diabetes being an autoimmune disease has been based is the detection of many different types of autoantibodies in patients with DM. These antibodies do not seem to play a major pathogenic role, at least as initial pathogenic insults; they seem to reflect the intensity of the underlying autoimmune reac-tion against the islet cell β cells. These antibodies can be detected before diabetes becomes clinically evident, and the number of different antibodies detected seems to be inversely correlated with the length of the disease-free interval in positive individuals. The following are the major types of autoantibodies detected in patients with IDDM.
1. Anti-islet cell antibodies (ICA) are classically detected by indirect immunofluo-rescence and react against membrane and cytoplasmic antigens of the islet cells. ICA are detected in as many as 90% of type 1 diabetic patients at the time of di-agnosis, but they diminish in frequency to 5–10% in patients with long-standing DM. Other interesting characteristics of ICA are their isotype distribution, with predominance of subclasses IgG2 and/or IgG4 (which have limited complement-activating properties), and their detection months or years before the appearance of clinical symptoms.
2. The best characterized islet cell antigens against which antibodies have been demonstrated in type 1 diabetics and individuals predisposed to develop the dis-ease are IA-2α and IA-2β (phogrin), two closely related β cell–associated tyro- sine phosphatases. About two thirds of the patients have antibodies to IA-2α , and about 60% of the patients have antibodies to IA-2 β . Epitope mapping studies suggest that the immunogenic epitopes are located in the intracytoplasmic seg-ment of these enzymes. Antibodies to glutamic acid decarboxylase (GAD) are also present in a large proportion of newly diagnosed diabetics (84%).
3. Anti-insulin autoantibodies are detected in as many as 92% of non–insulin-treated patients with IDDM at the time of diagnosis. The pathogenic significance of insulin autoantibodies is not clear, but the coexistence of anti-insulin antibod-ies and ICA has a strong predictive value for the future development of diabetes. Two theories have been proposed to explain the emergence of anti-insulin anti-bodies: (1) During destruction of the islet cells, insulin may be exposed to the im- mune system in a form that may be recognized as foreign. This could explain the development of anti-insulin antibodies in type I patients with insulitis (islet cell inflammation). (2) A recent finding of antigenic mimicry between insulin and a retroviral antigen, apparently leading to the spontaneous emergence of anti-in-sulin antibodies in non-obese diabetic prone mice, supports the alternative pos-sibility that anti-insulin antibodies may be triggered as a result of infection with an agent expressing cross-reactive antigen(s).
4. Induced anti-insulin antibodies can be found in all diabetic patients treated with insulin. The incidence of these antibodies was greater when bovine or porcine in-sulin was used. However, anti-human insulin antibodies can also be detected (less frequently) in patients treated with recombinant human insulin, whose ter-tiary configuration differs from that of the insulin released by the human pan-creas. The antibodies directed against therapeutically administered insulins ap-pear to be predominantly of the IgG2 and IgG4 isotypes.
Cell-mediated immunity is believed to have a more important pathogenic role in caus-ing islet cell damage than the autoantibodies to islet cells and insulin. One major argument in favor of the involvement of cell-mediated mechanisms is the fact that the pathological hallmark of recent onset diabetes is the mononuclear cell infiltration of the islet cells, known as insulitis. Similar observations can be made in animals with experimentally induced forms of diabetes. The predominant cells in the islet cell infiltrates are T lymphocytes, including both activated CD4+ and CD8+ T lymphocytes. Activated monocytes are also present in the infiltrates. The major pathogenic role seems to be played by CD4+ T cells, with a TH 0-TH1 cytokine secreting pattern. Those cells secrete large amounts of IL-2 and IFN- Îł.
The significance of increased IL-2 secretion may lie in the fact that IL-2 causes the upregulation of MHC-II in islet β cells, thus creating favorable conditions for the activa-tion of autoreactive cells. In experimental animal models, this change precedes the devel-opment of insulitis. IFN-γ , on the other hand, activates macrophages, causing the release of cytokines, such as IL-1 and IL-12, and toxic radicals. IL-1 has been shown to lead to β- cell damage by indirect mechanisms. IL-12 may promote the differentiation and activation of additional TH1 cells as well as the activation of cytotoxic T cells and NK cells. Toxic radicals, such as superoxide and nitric oxide, are known to be toxic to islet cells in vitro. It has been shown that IFN-γ can also activate the synthesis of oxygen active radicals and ni-tric oxide in β cells. These compounds can react with each other, forming peroxinitrate, which is highly toxic. Activated CD8+ cells may also be involved in monocyte activation through the secretion of IFN-γ . The main question that remains unanswered is the nature of the epitopes that are recognized by these cells and that trigger their activation.
Of crucial importance to our understanding of the pathogenesis of DM is the defini-tion of the insult(s) that may activate autoreactive T lymphocytes and trigger the disease. It is generally accepted that an environmental insult, most likely a viral infection, plays the initiating role, causing β-cell cytotoxicity. There is suggestive evidence supporting the pathogenic role of viral infections in some patient populations. For example, 12–15% of pa-tients with congenital rubella develop type 1 diabetes, particularly when they are DR3 or DR4 positive. But for the majority of diabetic patients, a link with any given viral infection remains elusive.
Whatever the initial insult to the β cell may be, it can be postulated that damaged β cells are ingested by macrophages, which will express islet cell–derived peptides in asso-ciation with MHC-II, thus creating conditions for the initiation of an autoimmune T-cell re-sponse. In addition, macrophages activated as a consequence of phagocytosis will release cytokines, such as IL-1 and IL-12, which activate T cells. Activated T cells, in turn, will re-lease IFN-γ , which activates macrophages and induces MHC-II expression. This mutual activation of macrophages and T cells results in the inflammatory process known as in-sulitis, which perpetuates β-cell destruction through the release of cytokines and toxic com-pounds and the differentiation of cytotoxic T cells. β-Cell death seems to result from both necrosis and apoptosis.
The nature of the epitopes recognized by autoreactive T cells has not been fully de-termined. Different authors have published results suggesting that insulin-derived, GAD-derived, and IA-2α and β epitopes are recognized by autoreactive T cells. The overall ev-idence suggests that the T-cell autoimmune response is polyclonal. Once the antigens recognized by sensitized T cells are identified, it will remain to be determined whether vi-ral infections play a pathogenic role, perhaps through upregulation of MHC-II and genera-tion of high concentrations of self peptides resulting from processing of proteins released by dying β cells.
2. Genetic Factors
Type 1 DM is a polygenic disease. Twenty different chromosomal regions possibly influ-encing the development of this form of diabetes have been identified. Of those, two have been better characterized.
The IDDM1 region, which includes the MHC genes determining resistance/suscep-tibility to diabetes, is considered to be the major genetic determinant of predisposition for the development of diabetes. Several DP and DQ alleles are associated with predisposition or resistance to diabetes. Ninety-five percent of diabetics express DR3 and/or DR4, com-pared to 42–54% of nondiabetics. This corresponds to a relative disease risk of 2–5. Sev-enty-five percent of diabetics express the Dw4 allele versus 54% of nondiabetics. Several DQ and DR alleles associated with susceptibility or resistance to diabetes have been iden-tified. In Caucasians, resistance is associated with the DQβw3.1 allele, characterized by the presence of aspartate (a negatively charged amino acid) in position 57 of the β chain. The presence of a neutral amino acid in that same position is characteristic of susceptibility al-leles, such as DQ 2 or DQ 8 (which are in linkage disequilibrium with DR3 and DR4), as is presence of arginine in position 52 of DQα . Individuals with two Asp-DQ alleles have the highest degree of predisposition to DM (Table 17.2).
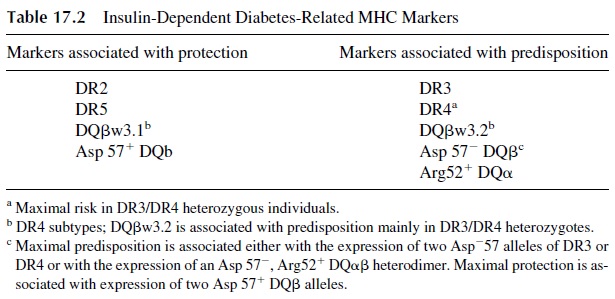
MHC-I genes associated with predisposition to develop diabetes have also been iden-tified. These MHC-I molecules are believed to be involved in the presentation of “diabeto-genic” peptides to CD8+ cells, as supported by the finding of activated CD8+ T cells in the infiltrates surrounding the islets. Two additional sets of genes may play significant roles in determining whether CD8+ T cells are activated or not:
1. Loci coding for proteasome components, which influence the type of peptides generated from autologous proteins. The generation of endogenous “diabeto-genic” peptides, therefore, may depend on the nature of proteasomes.
2. Loci coding for the synthesis of transport-associated proteins (TAP proteins) re-sponsible for the transport of endogenous peptides to the endoplasmic reticulum, where those peptides become associated with MHC-I peptides. The Tap genes are located in chromosome 6, near the MHC region, and may be transmitted in linkage disequilibrium with MHC-II genes. One Tap-2 gene allele (Tap-2*0101) is associated with susceptibility to diabetes (relative risk of 3.4), while another has been defined as protective in relation to diabetes.
The IDDM2 region includes the insulin gene and the insulin-growth factor II loci on chromosome 11. These genes have low expression in the thymus, perhaps impairing the ability to develop central tolerance to insulin-derived peptides.
Several other regions are linked to diabetes predisposition with variable consistency. These include regions with loci coding for insulin growth factor–binding proteins (chro-mosome 2), which appear to be linked with predisposition to develop diabetes, particularly in females, and the CTLA-4 gene (also in chromosome 2), which is likely to play a signif-icant role in the regulation of peripheral tolerance.
3. Genetic Factors in the Development of Diabetes
Several hypotheses have been advanced to explain how MHC molecules, proteasomes, and TAP proteins influence the development of diabetes, hinging on their ability to generate and present in association with MHC molecule β-cell–derived peptides involved in the elicitation of the autoimmune response resulting in diabetes (diabetogenic peptides). One of the many interpretations put forward is that the basis for the MHC-II role in protection or predisposition for type 1 DM is related to the differential ability to bind diabetogenic peptides generated from the ingestion of β cells undergoing spontaneous apoptosis and pre-sent them to the immune system. Protective MHC-II molecules would not bind those pep-tides, whereas predisposing MHC-II molecules would. Presentation of MHC-II–bound di-abetogenic peptides by itself is not sufficient to explain the development of autoimmunity, because in physiological conditions it is likely to result in tolerance. The emergence of IDDM seems to be preceded by an abnormally low expression of predisposing MHC-II molecules and co-stimulatory molecules by dendritic cells. As a consequence, the ability to maintain peripheral tolerance to diabetogenic peptides may be lost early in life.
Later on, as a consequence of strong activation secondary to a viral infection that causes β-cell damage and ingestion of damaged cells by activated APCs, these markers would be upregulated and their binding sites occupied with diabetogenic peptides gener-ated in the phagolysosomes where the β-cell debris is digested. The MHC-II–peptide com-plexes generated in this way should be expressed with sufficient density as to activate a vig-orous CD4+ T-cell response from individuals no longer tolerant. Activated CD8+ T cells are also present in the infiltrates surrounding the islets of diabetic patients and diabetic ex- perimental animals. Those cells must be activated by recognizing MHC-I–expressed dia-betogenic peptides, generated in patients with the proper conjunction of MHC-I molecules, proteasomes, and TAP proteins.
Finally, whether or not an individual carrying predisposing genes does or does not develop diabetes may depend on (1) whether all the cells necessary to start an active au-toimmune response receive the correct activation signals and (2) the lack of activation of counteregulatory cells that otherwise would keep the autoreactive clones in check.
4. Sequence of Pathogenic Events Leading to the Development of IDDM
Based on our current knowledge of the control of immunological responses and tolerance and on data accumulated from studies of IDDM patients and experimental animal models, the following hypothetical sequence of events leading to the development of IDDM can be proposed. First, one has to admit that autoreactive clones potentially able to be engaged in autoimmunity against pancreatic β cells persist in adult life. Under normal conditions, the interaction of those self-reactive TcR with MHC–diabetogenic peptide complexes ex-pressed in cells unable to deliver co-stimulatory signals results in weak, tolerogenic, or apoptotic signaling of the autoreactive T cells.
The structure of MHC molecules and the repertoire of proteasomes and TAP proteins genetically determine the level of expression of diabetogenic peptides. Those individuals able to express high levels of those peptides in association with MHC molecules are at greater risk to develop IDDM. A viral infection affecting the β cells or neighboring tissues leads to the activation of TH cells involved in the antiviral response. Those cells will deliver co-stimulatory signals to the autoreactive TH cells, pushing them into a state of activation rather than anergy. IL-2 and other cytokines released by activated T cells induce the ex-pression of MHC-II and CAMs in β cells.
The activated autoreactive T cells accumulate in the pancreatic islets and release chemotactic cytokines and interferon-Îł , which will attract and activate mono-cytes/macrophages to the area, where interactions with islet cells overexpressing CAMs will contribute to their fixation in the islets. The activated monocytes/macrophages release cytokines such as IL-1, IL-12, TNF, and toxic compounds such as oxygen active radicals and nitric oxide. The cytokines contribute to the damage by increasing the level of activa-tion of TH1 cells (IL-12), monocytes, and macrophages (IL-1, TNF). In addition, IL-1 and interferon-Îł (released by activated TH1 cells) induce the expression of Fas on islet cells. Islet cell death is a consequence of several mechanisms, including Fas-FasL apoptosis sig-naling and the release of toxic radicals that lead to oxidative changes of cell and organelle membrane lipids.
5. Immunotherapy
Two main approaches to immunotherapy have been evaluated: one involving the use of im-munosuppressants, the other involving tolerization.
Among immunosuppressants, cyclosporin A received considerable attention in trials, with the goal of preventing the full development of DM. To be effective, immunosuppres-sive therapy needs to be instituted in recently diagnosed patients with residual β-cell func-tion, but the treatment is only effective while cyclosporin A is administered. In the vast ma-jority of cases, progression to diabetes is seen soon after immunosuppressive therapy is discontinued.
Induction of tolerance seems possible after administration of insulin either by aerosol or by the oral route. Aerosol administration seems to result in the induction of γ/δ CD8+ T cells that secrete IL-10 and IL-4 and, in experimental animal models, are able to prevent the development of diabetes. Oral administration seems to generate a similar set of γ/δ CD4+ regulatory cells that secrete large amounts of TGF-β in addition to IL4 and IL-10. There are ongoing trials in which children identified as at risk by DQ haplotype analysis are given insulin by one of these routes, hoping to delay or prevent the onset of DM.
B. Acanthosis Nigricans
Acanthosis nigricans is a rare syndrome that received its name because of thickening and hyperpigmentation of the skin in the flexural and intertriginous areas. Patients with this dis-order develop a particularly labile form of diabetes associated with antibodies directed against the insulin receptor. These antibodies block the binding of insulin to the receptor. If the antibodies themselves are devoid of activating properties, they induce insulin-resis-tant diabetes. On the other side, the antibodies may stimulate the insulin receptor and cause hypoglycemia.
The clinical symptoms can be rather variable, depending on the biological properties of the predominant antibody population. Blocking antibodies to the insulin receptor cause hyperglycemia that does not respond to the administration of insulin (insulin-resistant dia-betes). In contrast, insulin receptor antibodies with stimulating properties may induce the cellular metabolic effects usually triggered by insulin, albeit in an abnormal and unregu-lated fashion. The clinical picture is one of hyperinsulinism. The same patient may undergo cycles of predominance of hypo- and hyperinsulinism-like symptoms, mimicking an ex-tremely brittle and difficult to control form of diabetes.
Related Topics