Chapter: Modern Pharmacology with Clinical Applications: Anticoagulant, Antiplatelet, and Fibrinolytic (Thrombolytic) Drugs
Anticoagulant Drugs
ANTICOAGULANT
DRUGS
Anticoagulant drugs inhibit
the development and en-largement of clots by actions on the coagulation phase.
They do not lyse clots or affect the fibrinolytic pathways.
Heparin
Two types of heparin are used
clinically. The first and older of the two, standard (unfractionated) heparin,
is an animal extract. The second and newer type, called low-molecular-weight
heparin (LMWH), is derived from unfractionated heparin.The two classes are
similar but not identical in their actions and pharmacokinetic characteristics.
Standard (Unfractionated) Heparin
Heparin (heparin sodium) is a mixture of highly elec-tronegative acidic
mucopolysaccharides that contain numerous N-
and O-sulfate linkages. It is
produced by and can be released from mast cells and is abundant in liver,
lungs, and intestines.
Mechanism of Action
The anticoagulation action of
heparin depends on the presence of a specific serine protease inhibitor (ser-pin) of thrombin,
antithrombin III, in normal blood.
Heparin binds to antithrombin
III and induces a con-formational change that accelerates the interaction of
antithrombin III with the coagulation factors. Heparin also catalyzes the
inhibition of thrombin by heparin co-factor II, a circulating inhibitor.
Smaller amounts of heparin are needed to prevent the formation of free thrombin
than are needed to inhibit the protease activ-ity of clot-bound thrombin.
Inhibition of free thrombin is the basis of low-dose prophylactic therapy.
Absorption, Metabolism, and Excretion
Heparin is prescribed on a
unit (IU) rather than mil-ligram basis. The dose must be determined on an
indi-vidual basis. Heparin is not absorbed after oral admin-istration and
therefore must be given parenterally. Intravenous
administration results in an almost immedi-ate anticoagulant effect. There
is an approximate 2-hour delay in
onset of drug action after subcutaneous admin-istration. Intramuscular
injection of heparin is to be avoided because of unpredictable absorption
rates, lo-cal bleeding, and irritation. Heparin is not bound to plasma proteins
or secreted into breast milk, and it does not cross the placenta.
Heparin’s action is
terminated by uptake and me-tabolism by the reticuloendothelial system and
liver and by renal excretion of the unchanged drug and its de-polymerized and
desulfated metabolite. The relative proportion of administered drug that is
excreted as un-changed heparin increases as the dose increases. Renal
insufficiency reduces the rate of heparin clearance from the blood.
Pharmacological Actions
The physiological function of
heparin is not com-pletely understood. It is found only in trace amounts in
normal circulating blood. It exerts an antilipemic effect by releasing
lipoprotein lipase from endothelial cells; heparinlike proteoglycans produced
by endothelial cells have anticoagulant activity. Heparin decreases platelet
and inflammatory cell adhesiveness to endothe-lial cells, reduces the release
of platelet-derived growth factor, inhibits tumor cell metastasis, and exerts
an an-tiproliferative effect on several types of smooth muscle.
Therapy with heparin occurs
in an inpatient setting. Heparin inhibits
both in vitro and in vivo clotting of blood. Whole blood clotting time and
activated partial thromboplastin time
(aPTT) are prolonged in propor-tion to blood heparin concentrations.
Adverse Effects
The major adverse reaction
resulting from heparin therapy is hemorrhage. Bleeding can occur in the
uri-nary or gastrointestinal tract and in the adrenal gland. Subdural hematoma,
acute hemorrhagic pancreatitis, hemarthrosis, and wound ecchymosis also occur.
The incidence of life-threatening hemorrhage is low but variable.
Heparin-induced thrombocytopenia of imme-diate and delayed onset may occur in 3
to 30% of pa-tients. The immediate type is transient and may not in-volve
platelet destruction, while the delayed reaction involves the production of
heparin-dependent an-tiplatelet antibodies and the clearance of platelets from
the blood. Heparin-associated thrombocytopenia may be associated with
irreversible aggregation of platelets (white clot syndrome). Additional
untoward effects of heparin treatment include hypersensitivity reactions (e.g.,
rash, urticaria, pruritus), fever, alopecia, hypoal-dosteronism, osteoporosis,
and osteoalgia.
Contraindications, Cautions, and Drug Interactions
Absolute contraindications
include serious or active bleeding; intracranial bleeding; recent brain, spinal
cord, or eye surgery; severe liver or kidney disease; dis-secting aortic
aneurysm; and malignant hypertension. Relative contraindications include active
gastrointesti-nal hemorrhage, recent stroke or major surgery, severe
hypertension, bacterial endocarditis, threatened abor-tion, and severe renal or
hepatic failure.
Drugs that inhibit platelet
function (e.g., aspirin) or produce thrombocytopenia increase the risk of
bleeding when heparin is administered. Oral
anticoagulants and heparin produce
synergistic effects. Many basic drugs
precipitate in the presence of the highly acidic heparin (e.g.,
antihistamines, quinidine, quinine, phenothiazines, tetracycline, gentamicin,
neomycin).
Heparin Antagonist
The specific heparin
antagonist protamine can be employed to neutralize heparin in cases of serious
hem-orrhage. Protamines are basic low-molecular-weight, positively charged
proteins that have a high affinity for the negatively charged heparin
molecules. The binding of protamine to heparin is immediate and results in the
formation of an inert complex. Protamine has weak an-ticoagulant activity.
Low-Molecular-Weight Heparin
Low-molecular-weight
fragments produced by chemical depolymerization and extraction of standard
heparin consist of heterogeneous polysaccharide chains of mo-lecular weight
2,000 to 9,000. The LMWH molecules contain the pentasaccharide sequence
necessary for binding to antithrombin III but not the 18-saccharide sequence
needed for binding to thrombin. Compared to standard heparin, LMWH has a 2- to
4-fold greater an-tifactor Xa activity than antithrombin activity.
LMWH has greater
bioavailability than standard heparin, a longer-lasting effect, and dose-independent
clearance pharmacokinetics. The predictable relation-ship between anticoagulant
response and dose allows anticoagulant control without laboratory tests. LMWH
is more effective than standard heparin in preventing and treating venous
thromboembolism. The incidence of thrombocytopenia after administration of LMWH
is lower than with standard heparin. Adverse drug reac-tions like those caused
by standard heparin have been seen during therapy with LMWH, and overdose is
treated with protamine.
LMWH is available for
subcutaneous administra-tion as enoxaparin (Lovenox),
dalteparin (Fragmin), ardeparin (Normiflo), and tinzaparin (Innohep). Dana-paroid (Orgaran), a heparinoid composed of
heparin sulfate, dermatan sulfate, and chondroitin sulfate, has greater factor
Xa specificity than LMWH. Bleeding due to danaparoid is not reversed by
protamine.
Orally Effective Anticoagulants
The orally effective
anticoagulant drugs are fat-soluble derivatives of 4-hydroxycoumarin or
indan-1,3-dione, and they resemble vitamin K. Warfarin is the oral anti-coagulant of choice. The indandione
anticoagulants have greater toxicity
than the coumarin drugs.
Mechanism of Action
Unlike heparin, the oral anticoagulants induce
hypoco-agulability only in vivo. They are vitamin K+ antagonists. Vitamin K+ is required to
catalyze the conversion of the precursors of vitamin K–dependent clotting
factors II, VII, IX, and X.This involves the posttranslational -car-boxylation
of glutamic acid residues at the N-terminal
end of the proteins. The -carboxylation step is linked to a cycle of enzyme
reactions involving the active hy-droquinone form of vitamin K+ (K1H2).The
regeneration of K1H1 by an epoxide reductase is blocked
by the oral anticoagulants. These drugs
thus cause hypocoagulabil-ity by inducing the formation of structurally
incomplete clotting factors.
Commercial warfarin is a
racemic mixture of S- and R-enantiomers; S-warfarin is more potent than
R-war-farin.
Absorption, Metabolism, and Excretion
Warfarin is rapidly and
almost completely absorbed af-ter oral administration and is bound extensively
( >95%) to plasma proteins. Since it is the unbound drug that produces the
anticoagulant effect, displace-ment of albumin-bound warfarin by other agents
may result in bleeding.Although these drugs do not cross the blood-brain
barrier, they can cross the placenta and may cause teratogenicity and
hemorrhage in the fetus.
Warfarin is inactivated by
hepatic P450 isozymes; hydroxylated metabolites are excreted into the bile and
then into the intestine. Hepatic disease may potentiate the anticoagulant
response.
Pharmacological Actions
Warfarin is used both on an
inpatient and outpatient basis when long-term anticoagulant therapy is
indi-cated. The onset of anticoagulation is delayed, the la-tency being
determined in part by the time required for absorption and in part by the
half-lives of the vitamin K–dependent hemostatic proteins. The anticoagulant ef-fect will not be evident in coagulation tests such
as pro-thrombin time until the normal factors already present in the blood are
catabolized; this takes 5 hours for factor VII and 2 to 3 days for prothrombin (factor II). The
an-ticoagulant effect may be preceded by a transient pe-riod of
hypercoagulability due to a rapid decrease in protein C levels. More rapid
anticoagulation is pro-vided, when necessary, by administering heparin.
Warfarin is administered in
conventional doses or minidoses to reduce bleeding. The dose range is ad-justed
to provide the desired end point.
Adverse Effects
The principal adverse
reaction to warfarin is hemor-rhage. Prolonged therapy with the coumarin-type
anti-coagulants is relatively free of untoward effects. Bleeding may be
observable (e.g., skin, mucous mem-branes) or occult (e.g., gastrointestinal,
renal, cerebral, hepatic, uterine, or pulmonary). Rarer untoward effects
include diarrhea, small intestine necrosis, urticaria, alopecia, skin necrosis,
purple toes, and dermatitis.
Contraindications, Cautions, and Drug Interactions
Oral anticoagulants are
ordinarily contraindicated in the presence of active or past gastrointestinal
ulcera-tion; thrombocytopenia; hepatic or renal disease; malig-nant
hypertension; recent brain, eye, or spinal cord sur-gery; bacterial
endocarditis; chronic alcoholism; and pregnancy. These agents also should not
be prescribed for individuals with physically hazardous occupations.
Minor hemorrhage caused by
oral anticoagulant overdosage can be treated by discontinuing drug
ad-ministration. Oral or parenteral vitamin K1 (phytona-dione)
administration will return prothrombin time to normal by 24 hours. This period
is required for de novo synthesis of biologically active coagulation factors. Serious hemorrhage may be stopped by
administration of fresh frozen plasma or plasma concentrates contain-ing
vitamin K–dependent factors.
Dietary intake of vitamin K+
and prior or concomi-tant therapy with a large number of
pharmacologically unrelated drugs can potentiate or inhibit the actions of oral
anticoagulants. Laxatives and mineral oil may re-duce the absorption of
warfarin. The patient’s pro-thrombin time and international normalized ratio (INR)
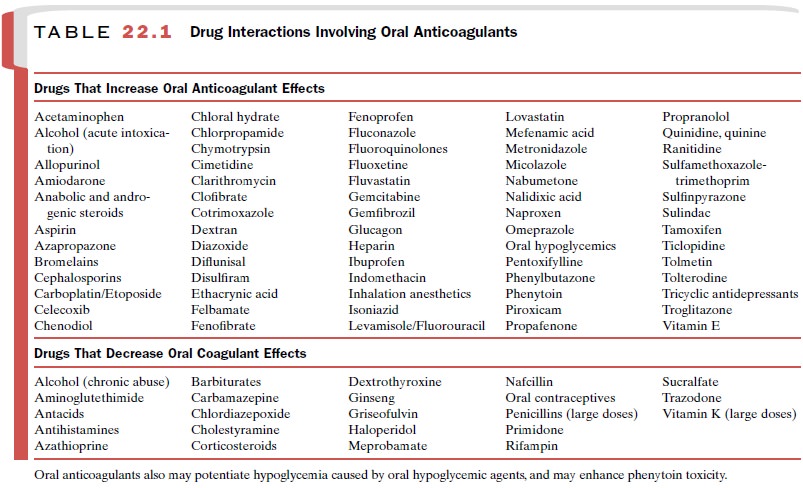
should be monitored when a drug is added or removed from therapy. Selected drug
interactions in-volving oral anticoagulants are summarized in Table 22.1.
Direct Thrombin Inhibitor Anticoagulants
Two drugs that are direct
inhibitors of thrombin but that do not involve antithrombin III or vitamin K+
in their mechanism of action have been approved to pro-vide intravenous
anticoagulation in patients with he-parin-induced thrombocytopenia. Lepirudin (Refludan) and bivalirudin (Angiomax), which are analogues of the
leech peptide anticoagulant hirudin, bind in a 1:1 com-plex with thrombin to
inhibit its protease activity. Argatroban (Acova,
Novastan), a synthetic analogue of arginine, interacts reversibly with and
inhibits throm-bin’s catalytic site. Both drugs have a short half-life.
Lipuridin is cleared following metabolism and urinary excretion of changed and
unchanged drug; hepatic me-tabolism of argatroban is a therapeutic advantage in
pa-tients with renal insufficiency. No antagonists for these drugs are
available.
Related Topics