Chapter: Modern Pharmacology with Clinical Applications: Metabolism and Excretion of Drugs
Renal Excretion
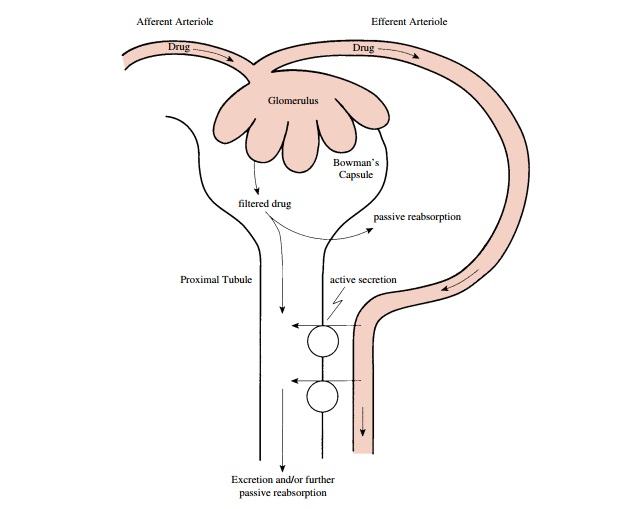
RENAL EXCRETION
Although some drugs are excreted through extrarenal pathways, the kidney is the primary organ of removal for most drugs (Figure 4.2), especially for those that are wa-ter soluble and not volatile. The three principal processes that determine the urinary excretion of a drug are glomerular filtration, tubular secretion, and tubular reab-sorption (mostly passive back-diffusion). Active tubular reabsorption also may have some influence on the rate of excretion for a limited number of compounds.
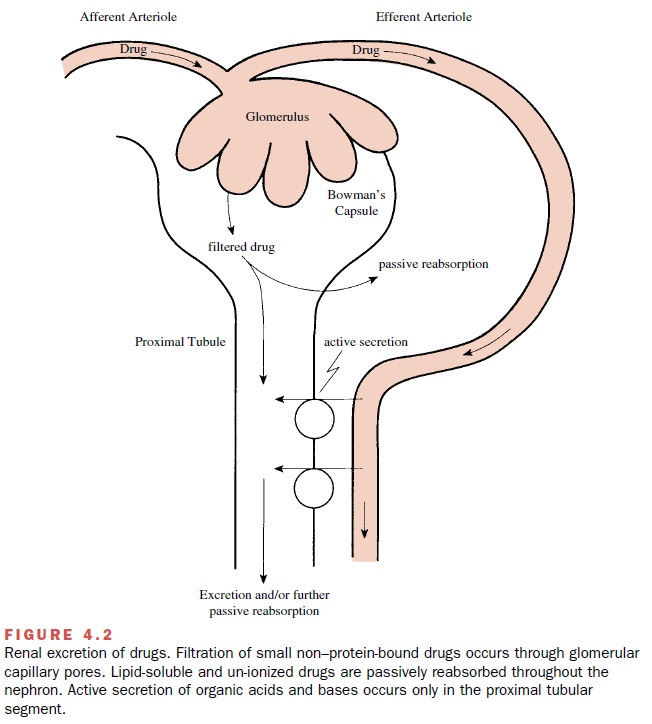
Glomerular Filtration
The ultrastructure of the
glomerular capillary wall is such that it permits a high degree of fluid
filtration while restricting the passage of compounds having relatively large
molecular weights. This selective filtration is im-portant in that it prevents
the filtration of plasma pro-teins (e.g., albumin) that are important for
maintaining an osmotic gradient in the vasculature and thus plasma volume.
Several factors, including
molecular size, charge, and shape, influence the glomerular filtration of large
mole-cules. The restricted passage of macromolecules can be thought of as a
consequence of the presence of a glomerular capillary wall barrier with uniform
pores.
Since approximately 130 mL of
plasma water is filtered across the porous glomerular capillary membranes each
minute (190 L/day), the kidney is admirably suited for its role in drug
excretion. As the ultrafiltrate is formed, any drug that is free in the plasma
water, that is, not bound to plasma proteins or the formed elements in the
blood (e.g., red blood cells), will be filtered as a result of the driving
force provided by cardiac pumping.
All unbound drugs will be
filtered as long as their mo-lecular size, charge, and shape are not
excessively large. Compounds with an effective radius above 20 Å may have their
rate of glomerular filtration restricted; hin-drance to passage increases
progressively as the molecu-lar radius increases, and passage approaches zero
when the compound radius becomes greater than about 42Å.
Charged substances (e.g., sulfated
dextrans) are usu-ally filtered at slower rates than neutral compounds (e.g.,
neutral dextrans), even when their molecular sizes are comparable. The greater
restriction to filtration of charged molecules, particularly anions, is
probably due to an electrostatic interaction between the filtered molecule and
the fixed negative charges within the glomerular capillary wall. These highly
anionic struc-tural components of the wall contribute to an electro-static
barrier and are most likely in the endothelial or glomerular basement membrane
regions.
Molecular configuration also
may influence the rate of glomerular filtration of drugs. Differences in the
three-dimensional shape of macromolecules result in a restriction of glomerular
passage of globular molecules (e.g., proteins) to a greater extent than of
random coil or extended molecules (e.g., dextrans). Thus, the effi-cient
retention of proteins within the circulation is at-tributed to a combination of
factors, including their globular structure, their large molecular size, and
the magnitude of their negative charge.
Factors that affect the
glomerular filtration rate (GFR) also can influence the rate of drug clearance.
For instance, inflammation of the glomerular capillaries may increase GFR and
hence drug filtration. Most drugs are at least partially bound to plasma
proteins, and therefore their actual filtration rates are less than the
theoretical GFR. Anything that alters drug–protein binding, however, will
change the drug filtration rate. The usual range of half-lives seen for most
drugs that are cleared solely by glomerular filtration is 1 to 4 hours.
However, considerably longer half-lives will be seen if extensive protein
binding occurs.
Also, since water constitutes
a larger percentage of the total body weight of the newborn than of
individu-als in other age groups, the apparent volume of distri-bution of
water-soluble drugs is greater in neonates. This results in a lower
concentration of drug in the blood coming to the kidneys per unit of time and hence
a decreased rate of drug clearance. The lower renal plasma flow in the newborn
also may decrease the glomerular filtration of drugs.
Passive Diffusion
An important determinant of
the urinary excretion of drugs (i.e., weak electrolytes) is the extent to which
sub-stances diffuse back across the tubular membranes and reenter the
circulation. In general, the movement of drugs is favored from the tubular
lumen to blood, partly because of the reabsorption of water that occurs
throughout most portions of the nephron, which results in an increased
concentration of drug in the luminal fluid. The concentration gradient thus
established will facilitate movement of the drug out of the tubular lu-men,
given that the lipid solubility and ionization of the drug are appropriate.
The pH of the urine (usually
between 4.5 and 8) can markedly affect the rate of passive back-diffusion.
Theback-diffusion occurs primarily in the distal tubules and collecting ducts,
where most of the urine acidification takes place. Since it is the un-ionized
form of the drug that diffuses from the tubular fluid across the tubular cells
into the blood, it follows that acidification in-creases reabsorption (or
decreases elimination) of weak acids, such as salicylates, and decreases
reabsorption (or promotes elimination) of weak bases, such as ampheta-mines.
However, should the un-ionized form of the drug not have sufficient lipid
solubility, urinary pH changes will have little influence on urinary drug
excretion.
Effects of pH on urinary drug
elimination may have important applications in medical practice, especially in
cases of overdose. For example, one can enhance the elimination of a
barbiturate (a weak acid) by adminis-tering bicarbonate to the patient. This
procedure alka-linizes the urine and thus promotes the excretion of the now
more completely ionized drug. The excretion of bases can be increased by making
the urine more acidic through the use of an acidifying salt, such as ammonium
chloride.
Active Tubular Secretion
A number of drugs can serve as substrates for the two active secretory systems in the proximal tubule cells. These transport systems, which actively transfer drugs from blood to luminal fluid, are independent of each other; one secretes organic anions (Figure 4.3), and the other secretes organic cations. One drug substrate can compete for transport with a simultaneously adminis-tered or endogenous similarly charged compound; this competition will decrease the overall rate of excretion of each substance. The secretory capacity of both the or-ganic anion and organic cation secretory systems can be saturated at high drug concentrations. Each drug will have its own characteristic maximum rate of secretion (transport maximum, Tm).
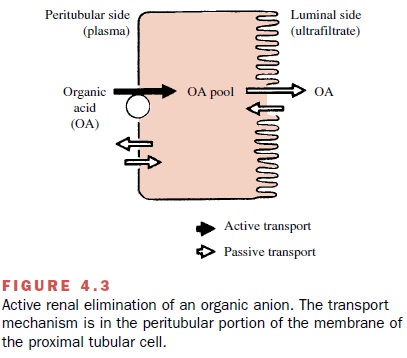
Some drugs that are not
candidates for active tubu-lar secretion may be metabolized to compounds that
are. This is often true for metabolites that are formed as a result of
conjugative reactions. Because the conju-gates are generally not
pharmacologically active, in-creases in their rate of elimination through
active secre-tion usually have little effect on the drug’s overall duration of
action.
These active secretory
systems are important in drug excretion because charged anions and cations are often
strongly bound to plasma proteins and therefore are not readily available for excretion by filtration. However,
since the protein binding is usually reversible, the active secretory systems
can rapidly and efficiently remove many protein-bound drugs from the blood and
transport them into tubular fluid.
Any drug known to be largely
excreted by the kid-ney that has a body half-life of less than 2 hours is
prob-ably eliminated, at least in part, by tubular secretion. Some drugs can be
secreted and have long half-lives, however, because of extensive passive
reabsorption in distal segments of the nephron. Several pharmacologically
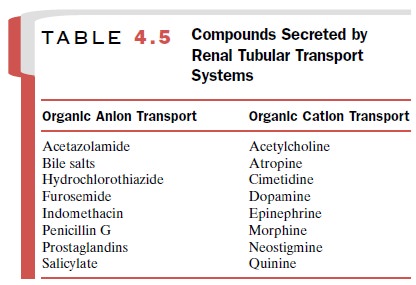
active drugs, both anions and cations, known to be secreted are listed in Table
4.5.
It is important to appreciate that these tubular transport mechanisms are not as well developed in the neonate as in the adult. In addition, their functional ca-pacity may be diminished in the elderly. Thus, com-pounds normally eliminated by tubular secretion will be excreted more slowly in the very young and in the older adult. This age dependence of the rate of renal drug se-cretion may have important therapeutic implications and must be considered by the physician who prescribes drugs for these age groups.
Finally, compounds that
undergo active tubular se-cretion also are filtered at the glomerulus (assuming
protein binding is minimal). Hence, a reduction in se-cretory activity does not
reduce the excretory process to zero but rather to a level that approximates
the glomerular filtration rate.
Active Tubular Reabsorption
Some substances filtered at
the glomerulus are reab-sorbed by active transport systems found primarily in
the proximal tubules. Active reabsorption is particularly important for
endogenous substances, such as ions, glucose, and amino acids (Fig. 4.4),
although a small number of drugs also may be actively reabsorbed. The probable
location of the active transport system is on the luminal side of the proximal
cell membrane. Bidirectional active
transport across the proximal tubule
also occurs for some compounds; that is, a drug may be both actively reabsorbed
and secreted. The oc-currence of such bidirectional active transport
mecha-nisms across the proximal tubule has been described for several organic
anions, including the naturally occurring uric acid . The major portion of filtered urate is probably reabsorbed,
whereas that eventually found in the urine is mostly derived from active
tubular secretion.
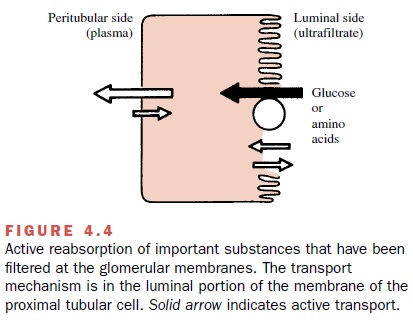
Most drugs act by reducing active transport rather than by enhancing it. Thus, drugs that promote uric acid loss (uricosuric agents, such as probenecid and sulfin-pyrazone) probably inhibit active urate reabsorption, while pyrazinamide, which reduces urate excretion, may block the active tubular secretion of uric acid. A com-plicating observation is that a drug may primarily in-hibit active reabsorption at one dose and active secre-tion at another, frequently lower, dose. For example, small amounts of salicylate will decrease total urate excretion, while high doses have a uricosuric effect.
This is offered as an explanation for the apparently
paradoxi-cal effects of low and high doses of drugs on the total excretory
pattern of compounds that are handled by re-nal active transport.
Clinical Implications of Renal Excretion
The rate of urinary drug
excretion will depend on the drug’s volume of distribution, its degree of
protein bind-ing, and the following renal factors:
Glomerular filtration rate
·
Tubular fluid pH
·
Extent of back-diffusion of the unionized form
·
Extent of active tubular secretion of the com-pound
·
Possibly, extent of active tubular reabsorption
Changes in any of these
factors may result in clini-cally important alterations in drug action. In the
final analysis, the amount of drug that finally appears in the urine will
represent a balance of filtered, reabsorbed (passively and actively), and
secreted drug. For many drugs, the duration and intensity of pharmacological
ef-fect will be influenced by the status of renal function, because of the
major role played by the kidneys in drug and metabolite elimination.
Ultimately, whether or not dosage adjustment (e.g., prolongation of dosing
inter-val, reduction in the maintenance dose, or both) be-comes necessary will
depend on an assessment of the degree of renal dysfunction, the percentage of
drug cleared by the kidney, and the potential for drug toxic-ity, especially if
renal function is reduced.
Biliary Excretion
The liver secretes about 1 L
of bile daily. Bile flow and composition depend on the secretory activity of
the he-patic cells that line the biliary canaliculi. As the bile flows through
the biliary system of ducts, its composi-tion can be modified in the ductules
and ducts by the processes of reabsorption and secretion, especially of
electrolytes and water. For example, osmotically active compounds, including
bile acids, transported into the bile promote the passive movement of fluid
into the duct lumen. In the gallbladder, composition of the bile is modified
further through reabsorptive processes.
The passage of most foreign
compounds from the blood into the liver normally is not restricted because the
endothelium of the hepatic blood sinusoids behaves as a porous membrane. Hence,
drugs with molecular weights lower than those of most protein molecules readily
reach the hepatic extracellular fluid from the plasma. A number of compounds
are taken up into the liver by carrier-mediated systems, while more lipophilic
drugs pass through the hepatocyte membrane by diffu-sion. The subsequent
passage of substances into the bile, however, is much more selective.
At least three groups of
compounds enter the bile. Compounds of group A are those whose concentration in
bile and plasma are almost identical (bile–plasma ra-tio of 1). These include
glucose, and ions such as NA+ , K+ , and Cl- .
Group B contains the bile salts, bilirubin glucuronide, sulfobromophthalein,
procainamide, and others, whose ratio of bile to blood is much greater than 1,
usually 10 to 1,000. Group C is reserved for com-pounds for which the ratio of
bile to blood is less than 1, for example, insulin, sucrose, and proteins.
Drugs can belong to any of these three categories. Only small amounts of most
drugs reach the bile by diffusion. However, biliary excretion plays a major
role (5–95% of the administered dose) in drug removal for some an-ions,
cations, and certain un-ionized molecules, such as cardiac glycosides. In
addition, biliary elimination may be important for the excretion of some heavy
metals.
Cardiac glycosides, anions,
and cations are trans-ported from the liver into the bile by three distinct and
independent carrier-mediated active transport systems, the last two closely
resembling those in the renal proxi-mal tubules that secrete anions and cations
into tubular urine. As is true for renal tubular secretion, protein-bound drug
is completely available for biliary active transport. In contrast to the bile
acids, the actively se-creted drugs generally
do not recycle, because they are not
substrates for the intestinal bile acid transport sys-tem, and they are
generally too highly charged to back-diffuse across the intestinal epithelium.
Thus, the ability of certain compounds to be actively secreted into bile
accounts for the large quantity of these drugs removed from the body by way of
the feces.
On the other hand, most drugs
that are secreted by the liver into the bile and then into the small intestine
are not eliminated through the feces. The physicochem-ical properties of most
drugs are sufficiently favorable for passive intestinal absorption that the
compound will reenter the blood that perfuses the intestine and again be
carried to the liver. Such recycling may continue (en-terohepatic cycle or
circulation) until the drug either un-dergoes metabolic changes in the
liver, is excreted by the kidneys, or both. This process permits the
conserva-tion of such important endogenous substances as the bile acids,
vitamins D3 and B12, folic acid, and estrogens (Table
4.6).
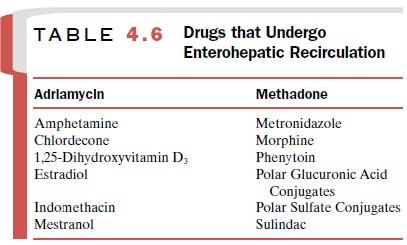
Extensive enterohepatic
cycling may be partly re-sponsible for a drug’s long persistence in the body.
Orally administered activated charcoal and/or anion ex-change resins have been
used clinically to interrupt en-terohepatic cycling and trap drugs in the
gastrointesti-nal tract.
As stated earlier, many foreign compounds are ei-ther partially or extensively metabolized in the liver.
Conjugation of a compound or
its metabolites is espe-cially important in determining whether the drug will
undergo biliary excretion. Frequently, when a com-pound is secreted into the
intestine through the bile, it is in the form of a conjugate. Conjugation generally en-hances biliary
excretion, since it both introduces a
strong polar (i.e., anionic) center into the molecule and increases its
molecular weight. Molecular weight may, however, be less important in the
biliary excretion of organic cations. Conjugated drugs will not be reab-sorbed
readily from the gastrointestinal tract unless the conjugate is hydrolyzed by
gut enzymes such as β-glucuronidase. Chloramphenicol glucuronide, for ex-ample, is
secreted into the bile, where it is hydrolyzed by gastrointestinal flora and
largely reabsorbed. Such a continuous recirculation may lead to the appearance
of drug-induced toxicity.
The kidney and liver are, in
general, capable of ac-tively transporting the same organic anion substrates.
However, the two organs have certain quantitative dif-ferences in drug affinity
for the transporters. It has been suggested that several subsystems of organic
anion trans-port may exist and that the binding specificities of the
transporters involved are not absolute but overlapping.
Liver disease or injury may impair bile secretion and thereby lead
to accumulation of certain drugs, for example
probenecid, digoxin, and diethylstilbestrol. Impairment of liver function
can lead to decreased rates of both drug metabolism and secretion of drugs into
bile. These two processes, of course, are frequently interrelated, since many
drugs are candidates for biliary secretion only after appropriate metabolism
has occurred.
Decreases in biliary
excretion have been demon-strated at both ends of the age continuum. For
example, ouabain, an unmetabolized cardiac glycoside that is se-creted into the
bile, is particularly toxic in the newborn. This is largely due to a reduced
ability of biliary secre-tion to remove ouabain from the plasma.
Increases in hepatic
excretory function also may take place. After the chronic administration of
either pheno-barbital or the potassium-sparing diuretic spirono- lactone, the
rate of bile flow is augmented. Such an in-crease in bile secretion can reduce
blood levels of drugs that depend on biliary elimination.
Finally, the administration
of one drug may influ-ence the rate of biliary excretion of a second
coadmin-istered compound. These effects may be brought about through an
alteration in one or more of the following factors: hepatic blood flow, uptake
into hepatocytes, rate of biotransformation, transport into bile, or rate of
bile formation. In addition, antibiotics may alter the intes-tinal flora in
such a manner as to diminish the presence of sulfatase and glucuronidase-containing
bacteria. This would result in a persistence of the conjugated form of the drug
and hence a decrease in its enterohepatic re-circulation.
Related Topics