Chapter: Medical Immunology: Primary Immunodeficiency Diseases
Humoral Immunodeficiencies
HUMORAL IMMUNODEFICIENCIES
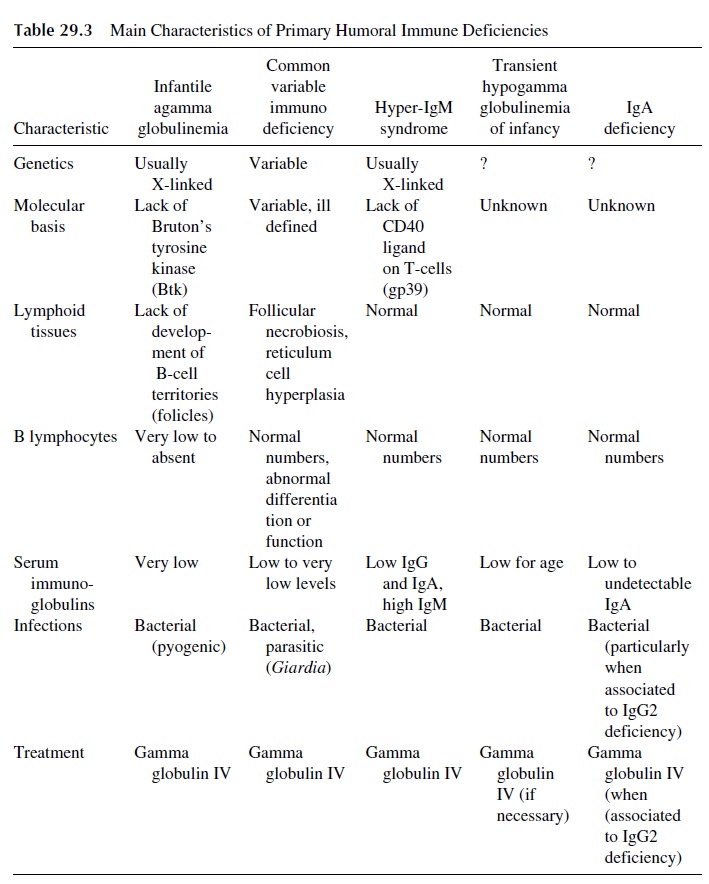
Humoral immunodeficiencies are those in which antibody synthesis is predominantly impaired. The general characteristics of the most important primary immunodeficiencies in-cluded in this group are summarized in Table 29.3. Patients with primary humoral immune deficiency generally do not develop clinical symptoms until after the first 6–12 months of life and the disappearance of maternally derived IgG. Most frequently these patients de-velop recurrent sinopulmonary infections with bacterial pathogens such as Haemophilusinfluenzae and Streptococcus pneumoniae. Deficiency in secretory IgA leads to chronicgastrointestinal illness with parasitic infections such as Giardia sp.

A. Transient Hypogammaglobulinemia of Infancy
This disorder is an accentuation of a normal physiological phenomenon. As maternal IgG is catabolized, with a half-life of approximately one month, infantile IgG levels are mainly dependent on the rate of antibody production.
Any delay in B-cell development results in low levels of immunoglobulins for age. All infants have lower levels of IgG and IgA when compared to adults. The nadir of IgG occurs between 3 and 6 months of age.
Most patients are seen because of an increased frequency and/or severity of bacterial infections. Low-for-age circulating immunoglobulin levels is the diagnostic hallmark. Dif-ferentiation with more severe forms of humoral immunodeficiencies is usually based on functional tests and enumeration of B cells.
Lymphocyte mitogenic responses and antibody response to challenge with toxoids are usually normal. Peripheral blood B lymphocytes are usually normal in number; in most cases, a deficiency of helper T-cell function appears to be responsible for the delay in immunoglobulin synthesis. Antigen-specific antibody pro-duction following infection or immunization is normal.
Most infants do not require therapy. Severe cases with recurrent infections can be treated with of intravenous gammaglobulin until the child’s immunoglobulin levels nor-malize. With time, most children will develop normal immune function.
B. Infantile Agammaglobulinemia (Bruton-Janeway Syndrome)
This is the prototype of “pure” B-cell deficiency. The disease is predominately transmitted as an X-linked trait. The defective gene is located on Xq21.2–22, the locus coding for the B-cell progenitor kinase or Bruton’s tyrosine kinase (Btk). Patients may have mutations at different sites within the locus resulting either in the lack of synthesis of the kinase or in the synthesis of an inactive form of the kinase.
Btk plays an important role in B-cell differentiation and maturation and is also part of the group of tyrosine kinases involved in B-cell signaling in adult life. Most mutations affecting Btk are associated with infantile agammaglobulinemia, but some patients with similar mutations have mild forms of immunodeficiency with variable levels of im-munoglobulin production. These findings suggest that B-cell differentiation may depend on additional, not yet identified cofactors.
Some forms of autosomal recessive infantile agammaglobulinemia have deletions of genes encoding parts of either the V region or of the Cµ region. Such deletions are associ-ated with a total lack of differentiation of B lymphocytes, similar to the defect seen in pa-tients with Btk deficiency.
1. Clinical Presentation
Infectious symptoms usually begin early in infancy as maternally acquired IgG disappears. The patients most commonly suffer from repeated infections caused by common pyogenic bacteria (Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae) of the sinopulmonary system. Pyoderma, purulent conjunctivitis, pharyngitis, purulent arthritis, otitis media, sinusitis, and bronchitis are common clinical findings. Severe life-threatening infections, such as pneumonia, empyema, meningitis, and septicemia, are also frequently seen. Chronic obstructive lung disease and bronchiectasis develop as a conse-quence of repeated bronchopulmonary infections in untreated older patients. Chronic diar-rhea and malabsorption caused by infections with Giardia lamblia are seen more frequently in these patients than in the general population. Arthritis of the large joints develops in about 30–35% of the cases and is sometimes associated with infection by Ureaplasma ure-alyticum. Agammaglobulinemic patients are at risk of developing paralytic polio after vac-cination with the attenuated polio vaccine. They also are at risk of developing chronic vi-ral meningoencephalitis, usually caused by echovirus.
2. Diagnosis
The diagnosis of agammaglobulinemia requires the quantitative assay of immunoglobulin levels. The serum levels for the three major immunoglobulins (IgG, IgA, and IgM) are greater than 2 SD below the normal level for children of the same age, although in infants low levels of IgG may be masked by the presence of maternal antibody. Usually the sum of the three major immunoglobulin isotypes is less than 100 mg/dL and electrophoresis fails to show a gamma globulin peak.
Definitive diagnosis requires at least one of the following: (1) detection of Btk mu-tations, (2) absence of Btk mRNA on Northern blot analysis of neutrophils or monocytes, (3) lack of Btk protein in monocytes or platelets, or (4) maternal male relatives with less than 2% CD19+ B cells.
The lack of B lymphocytes in peripheral blood ( <2% of the circulating lymphocytes are CD 19+ ) is one of the most important laboratory features of the disease, not shared by the most frequent cases of hypogammaglobulinemia, common variable immunodeficiency . Histological examination of lymphoid tissues shows lack of germinal centers and secondary follicles in lymph nodes and peri-intestinal lymphoid tissues. Plasma cells are absent both from peripheral lymphoid tissues and from bone marrow. Adenoids, ton-sils, and peripheral lymph nodes are hypoplastic. The thymus has normal structure, and the T-cell–dependent areas in peripheral lymphoid organs are normally populated. Normal numbers of B-cell precursors can be demonstrated in the bone marrow, indicating that the basic defect is a maturation block, restricted to B-cell development. Peripheral blood lym-phocyte counts are usually normal, T-lymphocyte counts are normal or elevated, T-lym-phocyte subsets are normal, and T-lymphocyte function is also normal.
Predictably, B-cell function is depressed. Isohemagglutinins are undetectable and the patients fail to produce antibodies after active immunization. Live, attenuated vaccines should be avoided in these patients.
3. Therapy
The primary strategy in therapy of infantile agammaglobulinemia is the prevention of in-fections. These children should not receive immunizations containing live, attenuated viruses (polio, measles, mumps, rubella, and varicella). Infections should be treated early and aggressively with antibiotics. Infections are best prevented by regular (monthly) infu-sions of intravenous gammaglobulin (IVIg). IVIg consists of purified polyclonal human IgG obtained from the pooled plasma of healthy blood donors. IgA and IgM, which can cause anaphylactoid reactions, are removed. Viruses that can be transmitted by blood transfusions are inactivated by detergent to minimize the risks of infection. IVIg penetrates mucosal sur-faces and crosses the blood-brain barrier poorly. Infections of the gastrointestinal tract some-times require the use of human or bovine colustrum or oral gamma globulin. Viral infections in the central nervous system may require infusions with intraventricular gamma globulin.
C. Common, Variable, Unclassified Immunodeficiency (“Acquired” Hypogammaglobulinemia)
This designation includes the most common form of hypogammaglobulinemia diagnosed in adults. The disorder is heterogeneous in presentation, with variable age of onset (usually after 2 years of age, most frequently between 15 and 35 years of age) and patterns of in-heritance. The clinical signs and symptoms are similar to X-linked agammaglobulinemia.
1. Physiopathology
A panel of experts who met under the auspices of the World Health Organization (WHO) in 1983 recognized several variants of common variable immunodeficiency.
1. Most variants of “acquired hypogammaglobulinemia” have normal or increased numbers of B lymphocytes in peripheral blood, but the B cells remain immature and do not respond adequately to in vivo stimulation.
2. T-cell function appears deficient in most cases, with abnormally low prolifera-tive responses to T-cell mitogens. T-cell receptor stimulation is followed by re-duced release of interleukins and reduced expression of CD40L (CD159). Thus, lack of proper T-cell help seems responsible for the lack of B-lymphocyte re-sponses.
3. In some patients the defect seems to result from excessive suppressor T-lym-phocyte activity.
2. Clinical Presentation
Sinopulmonary infections, primarily sinusitis, and bacterial pneumonia are the predomi-nant infections. Chronic obstructive pulmonary disease and bronchiectasis are frequent complications. Intestinal giardiasis is common and in some patients can lead to malab-sorption. Opportunistic infections involving P. carinii, mycobacteria, viruses, and other fungi are more frequent in these patients. Clinical features that differentiate common vari-able immunodeficiency from infantile agammaglobulinemia and other forms of hypogam-maglobulinemia are the increased incidence of autoimmune disease and malignancy among these patients. There is a high frequency of autoimmune cytopenias, pernicious anemia, arthritis, inflammatory arthritis, sprue, and polymyositis. Malignancy, particularly of the gastrointestinal lymphoid system, and nodular lymphoid hyperplasia at the intestinal tract are also common clinical manifestations.
3. Diagnosis
Serum immunoglobulin levels are variably depressed. In general the levels of IgG are 2 SD or more below the normal level for age. The aggregate level of the three major im-munoglobulin isotypes is usually below 300 mg/dL. The patients lack isohemagglutinins and fail to produce specific antibodies after immunization or infection. In contrast to what is observed in infantile agammaglobulinemia, these patients have, as noted, normal or in-creased numbers of B lymphocytes in peripheral blood, and those B cells can often be stimulated in vitro to produce immunoglobulins. Lymphoid tissues and tonsils, lymph nodes and spleen may be enlarged. Lymph node biopsies show morphological changes including necrobiosis of the follicles (also seen in the spleen) and/or reticular cell hyper-plasia (which may be the major contributing factor for the development of lym-phadenopathy and splenomegaly and in some patients seem to evolve into lymphoreticu-lar malignancies).
4. Treatment
Overall, therapy is similar to that for infantile agammaglobulinemia. Special emphasis should be directed toward preventing pneumonia and progression to bronchiectasis. IVIg should be administered at a dose and frequency to maintain total IgG levels of greater than 600 mg/dL at all times.
D. Selective Immunoglobulin A Deficiency
IgA deficiency is the most common immunodeficiency. The frequency estimates of IgA de-ficiency vary according to the criterion used to define it and with the sensitivity of the meth-ods used to measure IgA. An individual is considered IgA deficient when his or her con-centration of serum IgA is below 7 mg/dL, as measured by routine methods for immunoglobulin assay (such as radial immunodiffusion or immunonephelometry). De-fined by this criterion, IgA deficiency is diagnosed with a frequency of approximately 1 out of 600–800 normal Caucasian individuals.
1. Physiopathology
IgA deficiency appears to be a heterogeneous entity from the pathogenic point of view. In some cases phenotypic studies of circulating B cells show patterns similar to those of cord blood B lymphocytes, suggesting a differentiation abnormality, sometimes reflected by a defect in secretion of intracytoplasmic IgA. In other cases there is evidence for im-munoregulatory defects, such as:
· Predominant synthesis of IgG1 and IgG3 antibodies to pneumococcal polysaccha-rides, even when the serum levels of IgG2 are normal (IgM and IgG2 are the immunoglobulin isotypes of antipolysaccharide antibodies in humans).
· Longitudinal variations in IgA levels, which may fluctuate widely, from very low to normal and back to very low.
· Anti-IgA antibodies reacting with isotypic or allotypic determinants of IgA can be detected in about one third of the patients, usually in low titers. However, when present in high titers ( >80 when measured by passive hemagglutination), anti-IgA antibodies can cause hypersensitivity and possibly fatal reactions upon transfusion of IgA-containing blood products. Anti-IgA antibodies may con-tribute to the perpetuation of the IgA deficiency. The administration of radio-labeled IgA to patients with anti-IgA antibodies is followed by its rapid elimi-nation from the circulation (in a matter of hours). More significantly, a comparison of the levels of residual IgA in patients with and without anti-IgA antibodies demonstrated that those with antibodies have the lowest levels.
2. Clinical Presentation
Most cases of IgA deficiency are asymptomatic. Patients with combined IgA and IgG2 de-ficiency may present with recurrent sinus infections caused by bacteria with polysaccharide capsules. Infections withGiardia lamblia are more frequent in patients with IgA deficiency than in individuals with normal IgA levels. As in patients with agammaglobulinemia, this parasitic infection may lead to chronic diarrhea and malabsorption.
Abnormal immune reactivity is not unusual in IgA-deficient individuals. Many IgA-deficient individuals have antibodies to food proteins, which in most cases appear to be of no consequence. But IgA deficiency can also be associated with “autoimmune” disorders such as pernicious anemia and rheumatoid arthritis.
3. Diagnosis
Diagnosis is based on IgA assay. A patient older than 4 years with less than 7 mg/dL of serum IgA, normal levels of IgG and IgM, with no other primary or secondary immune de-ficiency fulfills the clinical and laboratory criteria for the diagnosis of selective IgA defi-ciency. Patients with other forms of primary immune deficiency such as ataxia-telangicta-sia can also have selective IgA deficiency.
4. Therapy
Treatment is usually targeted to relieve symptoms, using antibiotics as needed for infec-tions and treating allergies and sinus inflammation. Replacement therapy with IVIg for IgA deficiency is not recommended, because only small amounts of IgA are present in com-mercial gamma globulins and may cause adverse reactions. Administration of intravenous gamma globulin is indicated in patients with combined IgA and IgG2 deficiency or in IgA-deficient patients who fail to produce antibodies to bacterial polysaccharides. These pa-tients should receive IVIg preparations with very low IgA content to avoid hypersensitiv-ity reactions due to IgA antibodies.
IgA antibodies should be assayed in any known IgA-deficient patient considered for elective transfusion with IgA-containing blood products. If found, the blood bank should be notified so that steps can be taken to make sure that any blood transfused to the patient is IgA-depleted or a blood product or gamma globulin preparation depleted of IgA. IgA-depleted blood transfusions can be achieved by obtaining compatible blood from a healthy IgA-deficient donor or by extensively washing red cells to remove IgA-containing plasma. Patients should be educated about their increased risk of reactions to blood products.
E. X-Linked Hyper-IgM Syndrome
This syndrome is characterized by an inability of B cells to undergo immunoglobulin class switch resulting in low levels of IgG, IgA, and IgD in association to an elevation of IgM. In 70% of cases the disease is X-linked. Symptoms develop during the fist 5 years of life.
1. Genetics and Physiopathology
The X-linked hyper-IgM syndrome usually involves a mutation of the CD40 ligand (CD40L, CD159) gene, located on Xq26-27. As a consequence, T cells do not express CD40L and the signals mediated by CD40L-CD40 interactions are not delivered. This sig-nal is essential for B-lymphocyte differentiation and switching from IgM synthesis to the synthesis of other immunoglobulin classes. In addition to the failure to switch from IgM to IgG (IgA, IgE) synthesis during an immune response, germinal centers do not differentiate in the peripheral lymphoid tissues.
Other patients with hyper-IgM syndrome do express CD40L. In those patients, the molecular defect is believed to involve the second message systems, which transduce acti-vation signals after CD40L-CD40 interaction (mediated by the JAK-STAT and by the TRAF-MAK kinase pathways).
2. Clinical Presentation
These patients suffer from increased frequency of pyogenic infections, similar to those af-fecting patients with infantile agammaglobulinemia. There is also an associated finding of neutropenia that increases the predisposition for pyogenic and other opportunistic infec-tions. In addition to recurrent bacterial infections, patients carry an increased risk for Pneu-mocystis carinii pneumonia, cryptosporidium-related diarrhea, and aplastic anemia sec-ondary to parvovirus B19 infection. Autoantibodies are common. Other increased risks for these patients are lymphoproliferative syndromes, cancers of the biliary system, and biliary cirrhosis.
3. Diagnosis
The association of normal or high IgM levels with low IgG and IgA levels is a significant diagnostic clue. The numbers of T and B cells are normal or elevated. T-cell proliferative responses after mitogenic stimulation are normal, but the activated T cells do not express CD40L. B cells also respond to mitogenic stimulation, but produce IgM only. Definitive diagnosis of X-linked hyper-IgM syndrome requires either detection of a mutation in the CD40L gene or identification of maternal male relatives with confirmed diagnosis of this immunodeficiency.
4. Therapy
IVIg is used to correct the antibody deficiency associated with hyper-IgM syndrome. Pa-tients should be closely monitored for the development of autoimmunity and malignancy. Neutropenia has been corrected with the use of granulocyte colony-stimulating factors (G-CSF) and with bone marrow transplantation.
F. Antigen-Selective Immune Deficiencies
Those are immunodeficiencies in which the affected patients fail to produce antibodies fol-lowing a challenge with a specific antigen, while exhibiting normal immune responses to most other antigens.
1. Physiopathology
Two basic mechanisms can, at least theoretically, underlie antigen-specific immune defi-ciencies:
“Holes” in the immunoglobulin repertoire of the responding T-cell receptor implying that no binding sites for a given antigen are available either at the T- or at the B-cell level. Considering that immunogenic proteins are complex molecules with a variety of different epitopes, it is difficult to envisage how this mecha-nism could be involved. In the case of polysaccharides, simpler in structure and presenting a limited number of epitopes to the immune system, the hypothesis is more plausible.
Inefficient antigen presentation to helper T cells, implying that the nonresponse is a consequence of the lack of MHC-II molecules with adequate sites for binding of key peptides derived from antigen processing. This mechanism would apply only to T-dependent responses.
2. Clinical Presentation
Antigen-selective immune deficiencies are often undiagnosed and often asymptomatic. For example, using tetanus toxoid as immunogen, one can detect about 1 in 100 individuals whose humoral response is consistently low or undetectable, and the same is probably true with other immunogens. Patients who suffer from antigen-selective immune deficiencies and who have frequent infections are often misdiagnosed because most of the tests to eval-uate the immune system are normal. In some symptomatic cases, one or more of the IgG subclasses may be deficient. IgG2 deficiency can be associated with infections by polysac-charide-encapsulated bacteria such as Streptococcus pneumoniae and Haemophilus in-fluenzae.
3. Diagnosis
The diagnosis is eased on the failure to demonstrate the production of specific antibody fol-lowing challenge by infection or immunization.
4. Therapy
Replacement of antibody deficiency can be achieved with IVIg. Specific humanized mon-oclonal antibodies may be used to treat this disorder in the future
Related Topics