Chapter: Nutrition and Diet Therapy: Diet During Infancy
Special Considerations for Infants with Metabolic Disorders
SPECIAL CONSIDERATIONS
FOR INFANTS WITH METABOLIC DISORDERS
Some infants are born
with the inability to metabolize specific nutrients. These congenital
disabilities are called inborn errors of
metabolism. They are caused by mutations in the genes. There
is great variation in the seriousness of the conditions caused by these
defects. Some cause death at an early age, and some can be minimized so that
life can be supported by adjustments in the normal diet. Among children born
with these defects, there is, however, the common danger of damage to the
central nervous system because of their abnormal body chemistry. This results
in mental retardation and sometimes retarded growth. Early diagnosis of these
inborn errors, combined with diet therapy, increases the chances of preventing
retardation. Hospitals test newborns for some of these disorders as a matter of
course. If there is a family history of a certain genetic disorder, genetic
screening can be done. In addition, some of these abnormalities can be
discovered by amniocentesis.
Galactosemia
Galactosemia is a condition,
affecting 1 in 30,000 live births, in which thereis a lack of the liver enzyme transferase. Transferase normally
converts galac-tose to glucose. Galactose is the simple sugar resulting from
the digestion of lactose, the sugar found in milk. When transferase is missing
The newborn suffers diarrhea,vomiting, and edema,
and the child’s liver does not function normally. Cataracts may develop, galactosuria occurs, and mental
retardation ensues.
Diet Therapy.Diet therapy for galactosemia is the exclusion
of anythingcontaining milk from any mammal. During infancy, the treatment is
relatively simple because parents can feed the baby lactose-free, commercially
prepared formula and can provide supplemental minerals and vitamins. As the
child grows and moves on to adult foods, parents must be extremely careful to
avoid any food, beverage, or medicine that contains lactose. Nutritional
supplements of calcium, vitamin D, and riboflavin must be given so that the
diet is nutrition-ally adequate. This restricted diet may be necessary
throughout life, but some physicians allow a somewhat liberalized diet as the
child reaches school age. This may mean only small amounts of baked or
processed foods that contain small amounts of milk. Even this restricted diet
must be accompanied by careful and regular monitoring for galactosuria.
Phenylketonuria (PKU)
In phenylketonuria (PKU), infants lack the
liver enzyme phenylala-nine
hydroxylase, which is necessary for the metabolism of the amino acid phenylalanine. Infants seem to be
normal at birth, but if the disease is nottreated, most of them become
hyperactive, suffer seizures between 6 and 18 months, and become mentally
retarded. Public health law requires most hospitals today to screen newborns
for phenylketonuria. PKU babies typically have light-colored skin and hair.
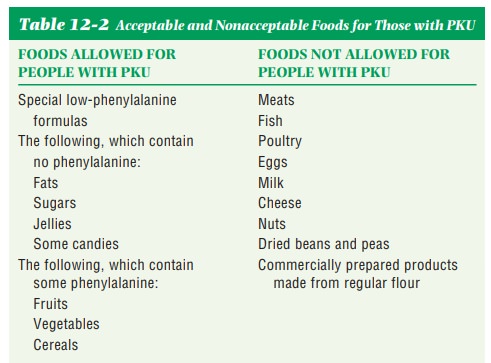
Diet Therapy.There is a special, nutritionally adequate,
commercial infantformula available for PKU babies. It is called Lofenalac. It has had 95% of
pheny-lalanine removed from its protein source. It provides just enough
phenylalanine for basic needs but no excess. The specific amount depends on the
infant’s size and growth rate. Regular blood tests determine the adequacy of
the amounts. Diets are carefully monitored for calorie and nutrient content and
are adjusted frequently as needs change. Except for fats and sugars, there is
some protein in all foods. Some of that protein is phenylalanine, so diets for
the growing child eating normal food must be carefully planned. There are two
varieties of synthetic milk available for older children. They are Phenyl-free and PKU-1, -2, or -3. None of
these contains any phenylalanine. They can be used as beverages or in puddings
and baked prod-ucts. Diets should be monitored throughout life to avoid mental
retardation and to control hyperactivity and aggressive behavior (Table 12-2).
Maple Syrup Urine Disease (MSUD)
Maple syrup urine disease (MSUD) is a congenital defect resulting in theinability to metabolize three amino acids: leucine, isoleucine, and valine. It is named for the odor of the urine of these infants and affects 1 in 100,000 to 300,000 live births. When the infant ingests food protein, there are increased blood levels of these amino acids, causing ketosis.
Hypoglycemia, apathy, and convulsions occur very
early. Depending on the extent of the disease, if not treated promptly, the
child can die from acidosis. Mild forms of the disease, if left untreated, will
cause mental retardation and bouts of acidosis.
Diet Therapy.The diet must provide sufficient calories and
nutrients butwith extremely restricted amounts of leucine, isoleucine, and
valine. A special formula and low-protein foods are used. Diet therapy appears
to be necessary throughout life.
Related Topics