Chapter: Essential Clinical Immunology: Experimental Approaches to the Study of Autoimmune Rheumatic Diseases
SjogrenŌĆÖs Syndrome: Epidemiology, Etiology, Pathogenesis, Clinical Features, Treatment, Animal Models
SJ├¢GRENŌĆÖS SYNDROME
Introduction and Epidemiology
Primary SjogrenŌĆÖs syndrome (SS) is a systemic autoimmune disease that targets the exocrine glands (Figure 11.3). It is character-izedbyxerostomia(drymouth),xerophthal-mia (dry eyes), and is usually accompanied by production of autoantibodies specific for the Ro RNA-binding protein. SS is a common autoimmune disorder. Prevalence estimates range from approximately 0.5 to 5 percent. Approximately, one-half of all cases of SS are primary and the remain-der occur as secondary SS. Secondary SS is defined as the former definition of primary Sj├ČgrenŌĆÖs in the presence of another auto-immune connective tissue disorder such as RA or SLE. Similar to most autoimmune disorders, most cases (approximately 90 percent) occur in women. The majority of cases occur in midlife; however, the disor-der is also seen in children and the elderly.
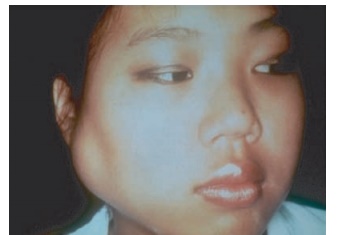
Figure 11.3 Parotid gland swelling in a patient with Sj├ČgrenŌĆÖs syndrome. ┬® 1972ŌĆō2004 American College of Rheumatology Clinical Slide Collection. Used with permission.
Etiology and Pathogenesis
Sj├ČgrenŌĆÖs syndrome has a strong immu-nogenetic component. Like many other autoimmune disorders, HLA studies have identified associations with MHC haplo-types. Studies have demonstrated that - DR2 and -DR3 associations in Sj├ČgrenŌĆÖs are secondary to linkage disequilibrium with HLA-DQ alleles. Genetically defined allelic markers have subsequently identified a large number of polymorphisms involving the HLA DRB1/DQA1/DQB1 haplotype, which adds to the complexity of the genetic background of SS. These polymorphisms have been shown to vary with ethnicity, clinical manifestations, and, importantly, with the autoantibody response. Similar to the situation in SLE, IFN-╬▒ has been impli-cated in SS pathogenesis based on expres-sion of interferon-induced genes in salivary gland tissue and increased type I interferon activity in serum.
Salivary gland pathology shows infil-tration with perivascular collections of T and B lymphocytes. The cells form dis-tinct aggregates, termed foci, in periduc-tal and periacinar locations that can result in replacement of epithelial structure. In rare cases, patients with SS develop non-HodgkinŌĆÖs B-cell lymphoma.
Clinical Features
The most common clinical manifestations of SS are ocular and oral. Patients most often perceive xerophthalmia as a foreign body-type sensation manifested by scratch-iness, grittiness, or irritation. Patients with xerostomia will complain of a parched sen-sation in the mouth, which often extends to the throat. Eating can be difficult without supplemental fluids. The reduction in sali-vary volume and loss of the antibacterial properties of saliva in the dry mouth accel-erate tooth decay. Unexplained rampant dental caries may, in fact, be the first sign of a dry mouth. Other xeroses such as dry skin and dry vaginal mucosa leading to irritation and dyspareunia may also occur.
Extraglandular manifestations of SS include musculoskeletal symptoms, with arthralgias and transient synovitis occur-ring in 54 to 84 percent of patients. Pulmo-nary involvement, which most commonly is cough due to xerotrachea, occurs in approximately 50 percent of patients. Mild autoimmune hepatitis has been reported to occur in approximately one-quarter of primary SS patients, with smooth muscle antibodies seen in 7 to 33 percent. Antimi-tochondrial antibodies have been reported in 7 to 13 percent of patients, suggesting an association of primary SS with primary biliary cirrhosis (PBC).
Neurologic disease is perhaps the most common significant extraglandular mani-festation of SS and can involve the cranial nerves, peripheral nerves, and rarely the central nervous system. Clinical reports suggest that half of SS patients have some form of neurological involvement, with estimates ranging from 22 to 76 percent.
Treatment of Sj├ČgrenŌĆÖs Syndrome
For the majority of patients, the main-stay of treatment for Sj├ČgrenŌĆÖs disease is external replacement of moisture. Patients should be encouraged to use tear substitutes often, and a variety of over-the-counter preparations are available. For xerostomia, artificial salivas are available, but these are generally not very success-ful and most patients find them unpalat-able. Patients whose symptoms of dryness are not optimally controlled by mois-ture replacement should be considered for treatment with secretory stimulants. There are two approved agents available for use as secretagogues in SS: pilocar-pine (Salagen) and cevemiline (Evoxac). Both agents have been shown, in con-trolled clinical trials, to increase salivary flow rate significantly in SS. There are no disease-modifying drugs that have been demonstrated to have specific efficacy for SS. Minor musculoskeletal symptoms usu-ally respond to NSAID therapy. Because erosive joint disease is rare, therapy with DMARDs is usually unnecessary; how-ever, hydroxychloroquine has been used to treat fatigue, arthralgia, and myalgia in primary SS. Cranial and peripheral neu-ropathy may be treated with low-dose tricyclic antidepressants or gabapentin. Symptomatic cases resistant to the previ-ously mentioned therapies may be treated with intravenous gammaglobulin. Central nervous system manifestations, which are felt to be due to Sj├ČgrenŌĆÖs, should be treated aggressively with high-dose oral or pulse intravenous corticosteroid and daily or monthly intravenous pulse cyclophosphamide.
Animal Models of Sj├ČgrenŌĆÖs Syndrome
Nonobese diabetic mice have been used predominantly as a model of autoimmune diabetes mellitus, but they also develop an autoimmune exocrinopathy characterized by hyposecretion of saliva and acinar cell atrophy.
MRL+/+ and MRL/lpr mice spontane-ously develop lacrimal and salivary gland inflammation and have been used as mod-els for SS.
IQI/Jic (IQI) mice are a model of pri-mary SS, unlike the animal models above that are autoimmune-prone mice that develop SS-like pathology associated with other autoimmune conditions, such as lupus, inflammatory arthritis, or insulitis. IQI mice spontaneously develop autoim-mune infiltration of lymphocytes into the lacrimal and salivary glands, leading to dacryoadenitis and sialoadenitis. The inci-dence of the disease is higher in females than in males. Sialoadenitis in female mice can be detected from two months of age onward and significant progression of the lesions is observed after nine months of age. Inflammatory lesions occurring at young ages mainly consist of CD4+ T cells with lesser abundance of CD8+ T cells, B cells, and macrophages, and the pro-portions of B cells and plasma cells are elevated in accordance with increasing magnification of the lesions. Production of ANAs, one of the prominent pathophysi-ological features in patients with SS, is also observed in old IQI mice. Moreover, IQI mice develop inflammatory lesions in the lung, pancreas, and kidney in addi-tion to the lacrimal and salivary glands that are similar to those in patients with primary SS. Thus, this murine model may be the most suitable for the investigation of the pathogenesis of SS, with progression from oral and ocular disease to a systemic disorder.
Related Topics