Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Regulatory Issues and Drug Product Approval for Biopharmaceuticals
Regulatory Routes - Biopharmaceuticals
REGULATORY ROUTES
From a regulatory perspective, a copy of a
biophar-maceutical product can be identified as a generic product. But in
practice it is unlikely, due to additional complexity, particularly safety
issues. The copy of a biopharmaceutical product is required to have a similar
safety and efficacy profile as the brand name/innovator product, and therefore
it is referred to as “biosimilar”. Biosimilar products are non-interchangeable.
According to the U.S. Food, Drug and Cosmetic Act,
the approval process can follow one of two routes: New Drug Application (NDA)
with two sub-classes, and Abbreviated New Drug Application (ANDA)
Section 505(b)(1): Full reports of investigations of
safety and efficacy are needed. This results in a full NDA, with right of
reference. This means that information about safety and efficacy can be used by
others to document safety and efficacy of the product.
Section 505(b)(2): This requires clinical studies,
without the right of reference, e.g., NDA for rDNA. Most of the biosimilar
products fall into this category.
Section 505(3): This is the route for ANDA
applications. This requires the dosage form to be pharmaceutically equivalent
and requires only bioequivalent studies. It does not require clinical or
pre-clinical studies. This is the route normally followed for small molecule
generic products.
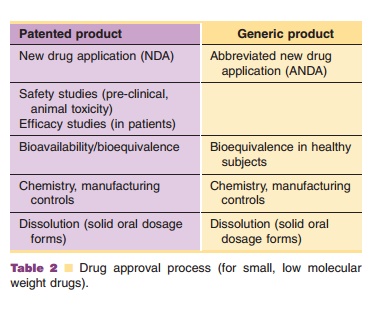
Biosimilar products are different from second
generation biopharmaceuticals (Table 2). The second generation
biopharmaceuticals have improved phar-macological properties/biological
activity compared to an already approved biopharmaceutical product which has
been deliberately modified. The second generation products are marketed with
the claim of improved clinical superiority. The second generation
biopharmaceuticals require a full New Drug Appli-cation and are not
interchangeable with the brand name product.
Two “types” of equivalence can be envisioned.
·
Within a
manufacturer—changes in the manufac-turing process, changes in the formulation
or a change in manufacturing site by a given manu-facturer. This requires
assurance of pharmaceutical equivalence. At times, depending upon the nature of
the change, a comparability study (see below) may be required. The FDA Guidance
on Com- parability is summarized in the insert box.
·
Between
manufacturers—This falls in the category of a biosimilar product and it
requires confirma-tion of pharmaceutical equivalence as well as bioequivalence.
Related Topics