Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Regulatory Issues and Drug Product Approval for Biopharmaceuticals
Regulatory Issues and Drug Product Approval for Biopharmaceuticals
Regulatory Issues and Drug
Product Approval for Biopharmaceuticals
INTRODUCTION
The term “biopharmaceuticals” is used to describe biotechnologically
derived drug products. Biopharma-ceuticals are protein-based macromolecules and
include, insulin, human growth hormone, the families of the cytokines and of
the monoclonal antibodies, antibody fragments, and nucleotide based systems
such as anti-sense oligonucleotides, siRNA and DNA preparations for gene
delivery. These are large complex molecules and are often heterogeneous
mixtures compared to synthetically manufactured, pure small molecules.
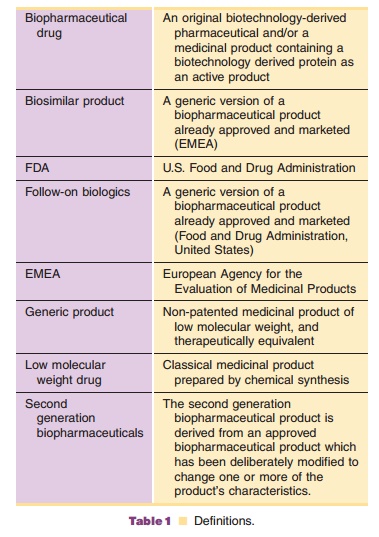
In the first years of the new millennium the regulatory landscape for
biopharmaceuticals changed. Before that time only original biopharmaceuticals
were approved by FDA and EMEA (Table 1) following the normal pathway of
approval including full scale clinical trials to ensure efficacy and safety and
this pathway still stands for original biopharmaceuticals. Then, a number of
biopharmaceutical drugs ran out of patent and the introduction of generic
versions of biopharmaceuticals became the center of lively debates: the new
products were called biosimilars (EMEA) or follow-on biologics (FDA) and issues
of debate were, for instance, structuring of the clinical trial programs. The
regulatory process for biosimi-lars/follow-on biologics is an evolutionary
process. Up until recently we have considered biopharmaceu-ticals as one
homogeneous class of compounds. But, that is an oversimplification. The
requirements for approval of biosimilar products should be based on the
structural complexity and clinical knowledge of and experience with the
reference biopharmaceutical product. The information provided here reflects the
status at December 2006.
Related Topics