Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Regulatory Issues and Drug Product Approval for Biopharmaceuticals
Background Regulatory Issues for Biopharmaceuticals
BACKGROUND
The aim of this chapter is to provide a comprehensive view on the
regulatory issues for the approval of a
biosimilar product. In order to have a better understanding of the regulatory
process involved, it is essential to appreciate the basic difference between
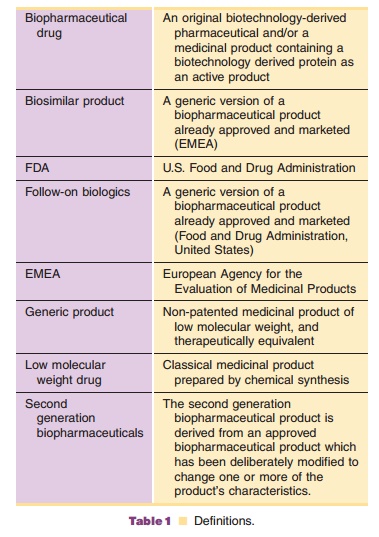
small drug molecules and macromolecules and their approval process. Table 1 provides
the definitions of different classes of medicinal drug products. Table 2
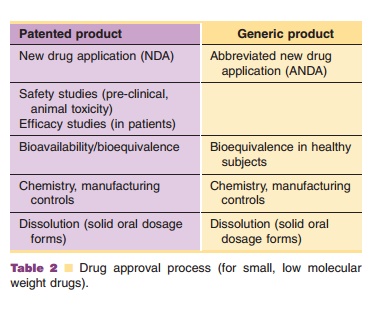
provides the steps involved in the drug approval process of small molecules.
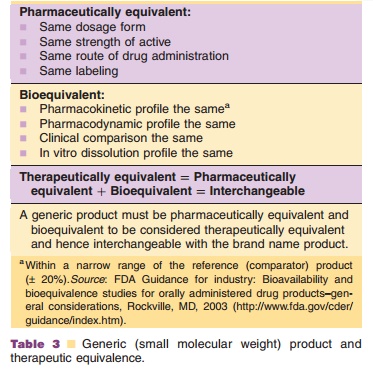
For the approval of a (small molecule) generic product, it must be
pharma-ceutically equivalent and bioequivalent (Table 3). Biopharmaceuticals
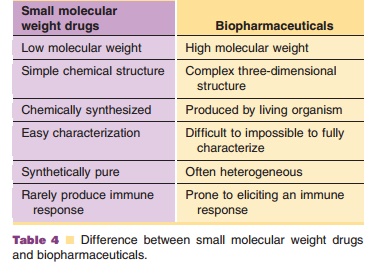
have a number of characteristics (Table 4) that set them aside from small
molecule drugs (Crommelin et al., 2003). The efficacy and safety of biotech
products depend on their complicated, rather labile shape built up of
secondary, tertiary and sometimes quaternary structures.
The mission of a regulatory authority is to “Assure that safe, effective
and high quality drugs are marketed in the country and are available to the
people”. Safety of an innovator’s drug product, be it a small molecule or
biopharmaceutical, is established through preclinical studies in animals and
controlled clinical studies in humans. Efficacy is established through clinical
studies in patients. A complete set of information on Chemistry, Manufacturing
and Controls (CMC-section) is needed in the dossier submitted for approval to
ensure that the drug substance and the drug product are, pure, potent and of
high quality. The CMC section should include full analytical characterization,
a description of the manufacturing process and test methods, and stability
data. In addition to the establishment of safety and efficacy of the drug
product, the approval process requires manufacturing of the drug product under
controlled current good manufacturing practice (cGMP) conditions. The cGMP
requirement ensures identity, potency, purity, quality and safety of the final
product. These features include process robustness and reproducibility,
validation, controls and testing. Batch release criteria are established to
assure complex manufacturing process of
biopharmaceuti-cals involving living organisms. The production process should
be controlled in every minor detail. Moreover, a long list of analytical
techniques is presented to characterize biopharma-ceuticals, but in many
instances full characterization is not possible with our current toolbox of
analytical techniques.
A generic product (small molecule) contains the same active ingredient
with inactive (most of the time) excipients as the reference product. As
explained above, this is not always possible in biopharmaceu-tical protein
products and therefore, these are not referred to as biogenerics. This biopharmaceutical
product contains the active ingredient which is similar (but not necessarily
equal) in characteristics to the reference product. For this reason, the
generic biopharmaceutical products are referred to as fol-low-on proteins or
follow-on biologics or biosimilar products. A biosimilar product has a
pharmacological and therapeutic activity that is similar to the reference
(comparator) product, but may differ in its pharma-ceutical equivalence
(characteristics). Biosimilar products are not generic products since it could
be expected that there may be subtle differences between the biosimilar and
reference product from the innovator and are not interchangeable with brand
name products.
Related Topics