Chapter: Essentials of Psychiatry: Psychiatric Pathophysiology: Dementia
Psychiatric Pathophysiology: Dementia
Psychiatric Pathophysiology: Dementia
Neuropathological Causes of Dementia in the Elderly
The most common neuropathological findings in
demented el-derly individuals are lesions associated with Alzheimer’s disease
(AD) and with cerebrovascular disease. Diseases such as Parkin-son’s disease,
diffuse Lewy body disease, Pick’s disease, and a constellation of heterogeneous
diseases collectively identified as frontotemporal dementia and characterized by
pronounced frontotemporal atrophy are also identified, albeit, less frequently
(Green et al., 2000; Xuereb et al., 2000; Hachinski and Munoz, 2000;
Perl, 2000; Dickson, 2001; Mckhan et al.,
2001). Often, AD and cerebrovascular disease, or AD and Parkinson’s
disease-like lesions (Perl et al.,
1998), are comorbid, making it difficult to ascribe dementia to either disease
entity alone and blurring the distinctions between them (Xuereb et al., 2000; Hachinski and Munoz, 2000;
Perl et al., 1998; Aguero-Torres and
Winbald, 2000; Snowdon et al., 1997;
Hardy and Gwinn-Hardy, 1999).
Neuropathology of Alzheimer’s Disease
In his original work, Alzheimer described the
plaques and tangles that have not only withstood modern scrutiny of the
original index case (Graeber and Mehraein, 1999) but have become the diagnostic
hallmarks of the disease known as Alzheimer’s disease. Neurofibrillary tangles
(NFT) and senile/neuritic plaques (NP) are the principal lesions of Alzheimer’s
disease (Rogers and Morrison, 1985; Tomlinson et al., 1970) and form the basis for the neuropathological
diagnosis of AD (Mirra et al., 1991;
Khachaturian, 1985; NIA-Reagan, 1997). In addition to NPs and NFTs, there is
generally macroscopic evidence of cerebral cortical atrophy, reduced brain
weight, and enlargement of the ventricles; however, because of the considerable
overlap between AD and normal control subjects and the frequency of similar
changes in other degenerative diseases (e.g., Pick’s disease), these
macroscopic findings are not diagnostically informative (Perl, 2000; Dickson,
2001; Adams and Duchen, 1992).
Neuritic plaques are extracellular spheroid
elements (10–120 µm in
diameter) with complex structures that include amyloid fibrils and dystrophic
neurites (Figure 14.1) that are frequently surrounded by microglia and reactive
astrocytes. The core of the plaque is composed of amyloid beta-peptide (Aβ) surrounded by abnormal neuronal
processes and fragments (Glenner and Wong, 1984; Selkoe, 1991, 1994, 2000). Aβ is derived from the sequential
proteolytic cleavage of a larger protein known as amyloid precursor protein
(APP). APP is highly conserved in evolution being expressed in all mammals
studied and in most organs. An above-normal accumulation of NPs with Aβ cores in the cerebral cortex,
hippocampal formation and subcortical structures has constituted the principal
diagnostic measure of AD (Mirra, 1991; Khachaturian, 1985).
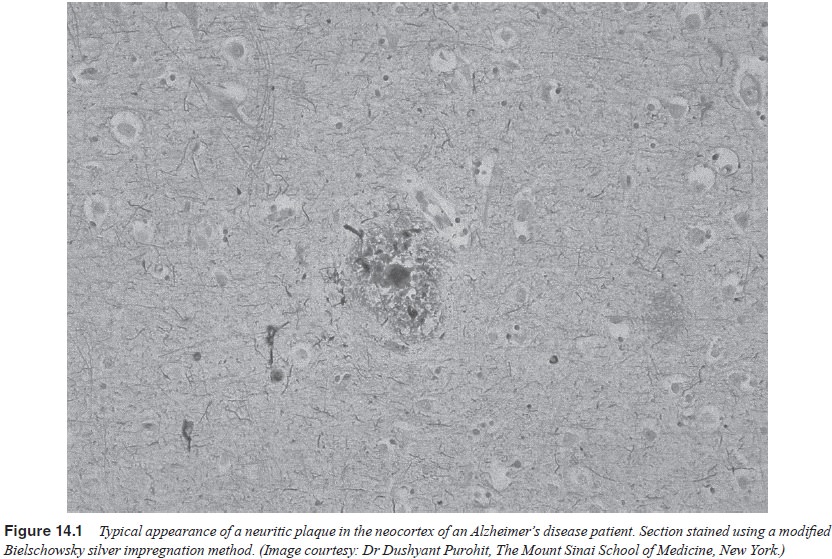
The precise mechanism(s) by which Aβ causes cell damage and neuronal
degeneration are not presently understood. It should be kept in mind, however,
that although it is broadly accepted that Aβ plays an
important role in AD, its role as the primary etiological agent in the disease
process is not universally accepted with cer-tainty (Davies, 2000).
Furthermore, how, if at all, NPs and NFTs interact and influence each other is
far from being understood.
Neurofibrillary tangles constitute the second
hallmark of AD. Although the most commonly used formal neuropathological
diagnostic criteria for AD rely almost exclusively on the density of
neocortical NPs (Mirra et al., 1991;
Khachaturian, 1985), a number of studies have argued that the development of
NFTs, especially in the entorhinal cortex, represents the earliest
neuropathological change in AD (Jellinger et
al., 1991; Braak and Braak, 1996b; Braak et al., 1996; Hyman, 1997; Bouras et al., 1993).
NFTs are intracellular coarse fibrillary structures
that can sur-round the neuronal nucleus and extend into the neuronal processes.
Although NFTs are observed in neurons that are otherwise appar-ently
“healthy’’, it is generally, but not universally assumed that NFT formation
leads to eventual neuronal death (Davies, 2000). NFTs are composed of pairs of
approximately 10 nm filaments entwined into helices with a signature
periodicity of approximately 160 nm.
Although neurons bearing NFTs in the hippocampus
and the entorhinal cortex are observed in many if not most elderly
indi-viduals, the “spread’’ of NFTs to the neocortex (Braak stage III and
above) is almost always associated with some level of dementia.
Despite considerable advances in understanding the
neuro-biology and genetics of Aβ and NFTs
(see later), linkage between these two apparently diverse lesions has not been
clearly estab-lished. It may be tempting to argue that AD is the result of the
coincidence of two relatively independent neurobiological abnor-malities. It is
well established that a single mutation in the APP or an APP-related gene can
cause AD. Thus, there are considerable gaps in our knowledge of the mechanisms
by which the full spec-trum of AD neuropathology is expressed and a more
complete understanding of the etiology of AD must await further research.
Neurochemical Deficits in Alzheimer’s Disease
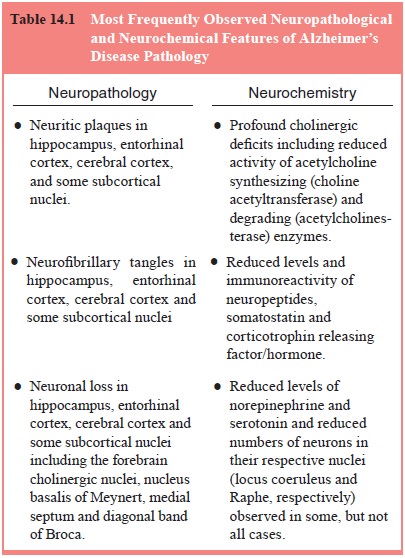
The neuropathology of AD is accompanied with significant neurochemical pathology and dysfunction. A central tenet of Alzheimer’s disease, established 20 years ago and repeatedly replicated, is the loss of cortical cholinergic markers, specifically the reduction of the activities of the enzymes choline acetyltrans-ferase (ChAT) and acetylcholinesterase (AChE), in postmortem tissue from Alzheimer’s patients (Perry et al., 1977; Davies and Maloney, 1976; Bowen et al., 1982). This abnormality has been shown to correlate with neuropathological markers and with the severity of dementia (Perry et al., 1978; Bierer et al., 1995a). Other neurochemical abnormalities that exist in the AD brain include loss of corticotropin releasing factor (CRF) (De Souza et al., 1986; Nermeroff et al., 1991) and somatostatin immunoreac-tivity (Davies and Terry, 1981; Davies et al., 1980), and decreases in noradrenergic (Adolfsson et al., 1979; Gottfries et al., 1989; Chan-Palay and Asan, 1989; Teicher et al., 1988), serotonergic (Cross et al., 1986a, 1986b; Gottfries, 1990), GABAergic (Ellison et al., 1986; Sasaki et al., 1986; Simpson et al., 1988; Chu et al., 1987a, 1987b; Hardy et al., 1987) and glutamatergic (Ellison et al., 1986; Kowall and Beal, 1991; Chalmers et al., 1990) mark-ers in some patients. Table 14.1 summarizes some of the principal neuropathologic and neurochemical findings in AD.
Synapses and Synaptic Proteins in Alzheimer’s Disease
Given the neuronal degeneration and the reductions in neuronal size and arborization in AD, it is only reasonable that the numbers of synapses and their integrity should be compromised in AD. Synaptic neurotransmission, like all forms of exocytosis, requires an array of proteins. These proteins have varying ion and guanine nucleotide sensitivities, and possess intricate functional and biochemical kinships. Synaptic proteins on vesicles within the synapse and on synaptic membranes interact to enable neurotransmitter-bearing vesicles to dock with synaptic membranes and become ready for neurotransmitter release. Although losses of synaptic proteins are not unexpected in a degenerative disease, the fact that the loss of synaptic marker proteins significantly and profoundly correlate with the severity of dementia (Terry et al., 1991; Masliah et al., 1989; Lassmann et al., 1993; Heinonen et al., 1995; Tiraboschi et al., 2000) suggest that the loss of synapses may play a pivotal role in the expression of the symptoms of AD.
Inflammatory Processes in Alzheimer’s Disease
The identification or recognition of
neurobiological indices of inflammation in the AD brain was initially
controversial because of the belief that the brain was immunologically
privileged (Rogers and Shen, 2000). More than a decade of research and hundreds
of studies have now confirmed the presence of inflammatory responses in the AD
brain (Akiyama et al., 2000) and have
raised the possibility of the potential benefit of anti-inflammatory
therapeutic avenues (Akiyama et al.,
2000; Aisen, 2000; Anthony et al.,
2000). It is generally acknowledged that inflammation of the brain or its neurons is unlikely to be the primary etiological
factor in AD, but that it is initiated by AD-specific pathogenic processes such
as deposition of NPs and/or NFTs. However, as in many other diseases, once
initiated, inflammatory responses may contribute to the progression of the
disease and its malignancy (Rogers and Shen, 2000).
Genetic Factors in Alzheimer’s Disease
Much has been learned regarding the contribution of
genetic factors to AD over the past two decades (George-Hyslop, 2000a, 2000b).
However, the impact of these genetic contributors to dementia severity is not
yet well understood. Significant evidence has accumulated for genetic traits
that confer susceptibility to AD. Four genes, beta-amyloid precursor protein (β-APP) (Selkoe, 1997, 2000b;
George-Hyslop et al., 1987; Robakis et al., 1987; Goate et al., 1991), presenilin-1 (PS1) (Selkoe, 1997, 2000b; Robakis et al., 1987; Sherrington et
al., 1995), presenilin-2 (PS2) (Selkoe, 1997, 2000b; Robakis et al., 1987; Rogaev et al., 1995), and apolipoprotein E
(APOE) (Selkoe, 1997, 2000b; Robakis et al., 1987; Saunders et al., 1993; Poirier, 2000) are of
particular note. The fact that the three most prominent AD-associated mutations
all affect APP processing, albeit by different mechanisms, increase Aβ production, strongly suggests
that Aβ is a prominent pathogenetic
agent in AD and strongly supports the amyloid hypothesis of AD (George-Hyslop,
2000a, 2000b), especially since emerging data suggest that even the epsilon-4
allele of APOE may influence Aβ
processing.
Although a thorough understanding of the
development of different neuropathological features of AD and their
relationship to the progression of dementia must await the development of
sensitive in vivo markers of
neuropathology, the existing evidence supports the hypothesis that NPs and NFTs
are “dose-depend-ently’’ associated with the earliest manifestations of AD-type
dementia and that inflammatory responses and neurochemical deficits such as the
cholinergic deficit become evident as demen-tia severity becomes more
pronounced.
Dementia in Schizophrenia and its Neuropathological and Neurochemical Sequelae
Chronic schizophrenia, with its onset in early
adulthood can in geriatric years become a disorder that is commonly associated
with dementia (Davidson and Haroutunian, 1995; Harvey et al., 1992, 1996; Davidson et
al., 1995). The findings of continuing deterioration of the symptoms of
schizophrenia, the cognitive performance of schizophrenics, especially the
apparent dementia that has been documented in elderly schizophrenics (Davidson
and Haroutunian, 1995; Harvey et al.,
1997, 1999a, 1999b;), and progressive enlargement of the ventricles, especially
in poor-outcome schizophrenics (Davis et
al., 1998) has raised the possibility of a neurodegenerative component to
the disease process or to its dementia.
The age-related emergence of cognitive impairment
in schizophrenia fueled hypotheses linking dementia in elderly schizophrenics
to AD and positing that the dementia of late-life schizophrenia was a
consequence of comorbid AD (Davidson and Haroutunian, 1995; Arnold et al., 1994). However, several large
postmortem studies failed to support this hypothesis. Thus, although there is
clear evidence for dementia in at least a subpopulation of elderly
schizophrenics, the neuropathological and neurobiological substrates of this
dementia have remained elusive and, as of yet, undetermined.
Related Topics