Chapter: Medical Surgical Nursing: Management of Patients With Immunodeficiency
Phagocytic Dysfunction - Primary Immunodeficiencies
Primary Immunodeficiencies
Primary
immunodeficiencies, rare disorders with genetic origins, are seen primarily in
infants and young children. To date, more than 95 immunodeficiencies of genetic
origin have been identi-fied (Buckley, 2000). Symptoms usually develop early in
life after protection from maternal antibodies decreases. Without treat-ment,
infants and children with these disorders seldom survive to adulthood. These
disorders may involve one or more components of the immune system. Symptoms of
immune deficiency diseases are related to the role that the deficient component
normally plays (Table 51-1).

PHAGOCYTIC
DYSFUNCTION
Pathophysiology
A variety of primary defects of phagocytes
may occur; nearly all of them are genetic in origin and affect the innate
immune system. In some types of phagocytic disorders, the neutrophils are
im-paired so that they cannot exit the circulation and travel to sites of
infection. As a result the patient cannot mount a normal inflam-matory response
against pathogenic organisms (Lekstrom-Himes Gallin, 2002). In some disorders, the
neutrophil count may be very low; in others, it may be very high because the
neutrophils re-main in the vascular system.
Clinical Manifestations
Phagocytic cell disorders are manifested by
an increased incidence of bacterial and fungal infections due to normally
nonpathogenic organisms (Lekstrom-Himes & Gallin, 2002). People with these
disorders may also develop fungal infections from Candida or-ganisms and viral
infections from herpes simplex or herpes zoster virus. These patients
experience recurrent furunculosis, cutaneous abscesses, chronic eczema,
bronchitis, pneumonia, chronic otitis media, and sinusitis. In one type of phagocytic
disorder, hyper-immunoglobulinemia E (HIE) syndrome, formerly known as Job
syndrome, white blood cells cannot produce an inflammatory re-sponse to the
skin infections; this results in deep-seated cold ab-scesses that lack the
classic signs and symptoms of inflammation (redness, heat, and pain).
While patients with phagocytic cell disorders
may be asympto-matic, severe neutropenia may be accompanied by deep and painful
mouth ulcers, gingivitis, stomatitis, and cellulitis. Death from overwhelming infection
occurs in about 10% of patients with severe neutropenia. Chronic granulomatous
disease, another type of primary phagocytic disorder, produces recurrent or
persistent infections of the soft tissues, lungs, and other organs; these are
re-sistant to aggressive treatment with antibiotics (Lekstrom-Himes &
Gallin, 2002).
Assessment and Diagnostic Findings
Diagnosis is based on the history, signs and
symptoms, and lab-oratory analysis of the cytocidal (causing the death of
cells) activ-ity of the phagocytic cells by the nitroblue tetrazolium reductase
test. A history of recurrent infection and fever in a child and occasionally in
an adult is an important key to the diagnosis. Failure of an infection to
resolve with usual treatment is also an important indicator (Lekstrom-Himes
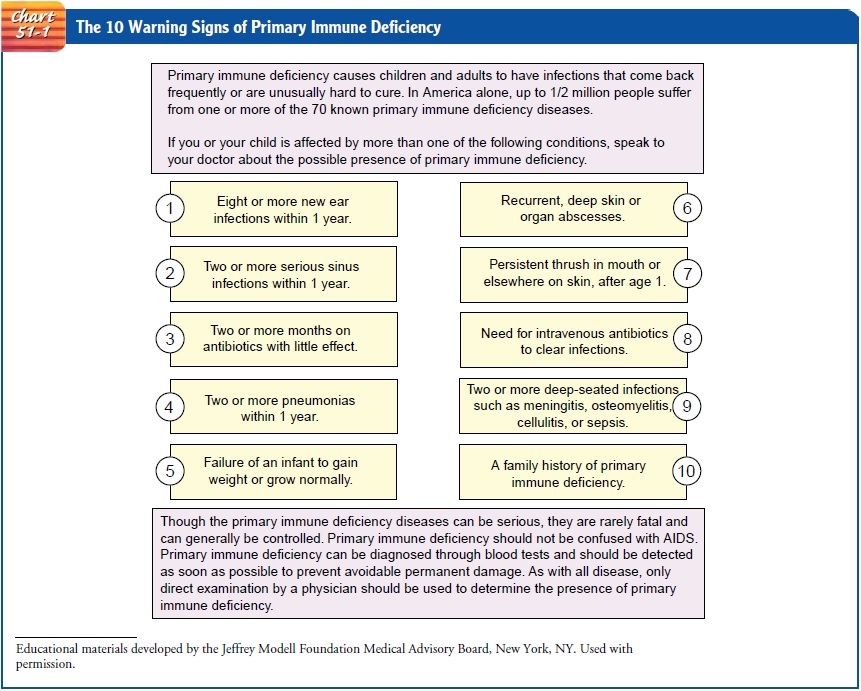
& Gallin, 2002). See Chart 51-1 for a summary of warning signs of primary
immunodeficiency disorders.
Medical Management
Because the signs and symptoms of infection are often blunted because of an impaired inflammatory response, early diagnosis and treatment of infectious complications can be lifesaving (Lekstrom-Himes & Gallin, 2002). Management of phagocytic cell disorders includes treating bacterial infections with prophy-lactic antibiotic therapy. Additional treatment for fungal and viral infections is often needed. Granulocyte transfusions, although used, are seldom successful because of the short half-life of the cells. Treatment with granulocyte-macrophage colony-stimulating factor (GM-CSF) or granulocyte colony-stimulating factor (G-CSF) may prove successful because these proteins draw nonlymphoid stem cells from the bone marrow and hasten their maturation.
Related Topics