Chapter: Clinical Anesthesiology: Clinical Pharmacology: Hypotensive Agents
Nitrovasodilators: Sodium Nitroprusside
Nitrovasodilators
SODIUM NITROPRUSSIDE
Mechanism of Action
Sodium nitroprusside and other
nitrovasodilators relax both arteriolar and venous smooth muscle. Its primary
mechanism of action is shared with other nitrates (eg, hydralazine and
nitroglycerin). As these drugs are metabolized, they release nitricoxide, which activates guanylyl
cyclase. This enzymeis responsible for the synthesis of cyclic guanosine
3',5'-monophosphate (cGMP), which controls the phosphorylation of several
proteins, including some involved in the control of free intracellular calcium
and smooth muscle contraction.
Nitric oxide, a naturally occurring
potent vasodilator released by endothelial cells (endothe-lium-derived relaxing
factor), plays an important role in regulating vascular tone throughout the
body. Its ultrashort half-life (<5 s) provides
sensi-tive endogenous control of regional blood flow.Inhaled nitric oxide is a
selective pulmonary vasodilator that is beneficial and routinelyused in the
treatment of reversible pulmonary hypertension.
Clinical Uses
Sodium nitroprusside is a potent and
reliable antihy-pertensive. It is usually diluted to a concentration of 100
mcg/mL and administered as a continuous intra-venous infusion (0.5–10 mcg/kg/min).
Its extremely rapid onset of action (1–2 min) and fleeting dura-tion of action
allow precise titration of arterial blood pressure. A bolus of 1–2 mcg/kg
minimizes blood pressure elevation during laryngoscopy but can cause transient
hypotension in some patients. Thepotency of this drug requires frequent blood
pres-sure measurements—or, preferably, intraarterial monitoring—and the use of
mechanical infusion pumps. Solutions of sodium nitroprusside must be protected
from light because of photodegradation.
Metabolism
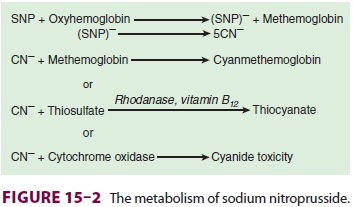
After parenteral injection, sodium
nitroprus-side enters red blood cells, where it receives an electron from the
iron (Fe2+) of oxyhemoglobin. This nonenzymatic
electron transfer results in an unstable nitroprusside radical and methemoglo-bin
(Hgb Fe3+). The former moiety spontaneously
decomposes into five cyanide ions and the active nitroso (N == O) group.
The cyanide ions can be involved in one
of three possible reactions: binding to methemoglobin to form cyanmethemoglobin; undergoing a
reaction in the liver and kidney catalyzed by the enzyme rho-danase
(thiosulfate + cyanide → thiocyanate); or binding to tissue
cytochrome oxidase, which inter-feres with normal oxygen utilization ( Figure 15–2).The
last of these reactions is responsible for the development of acute cyanide toxicity,characterized by
metabolic acidosis, cardiac arrhyth-mias, and increased venous oxygen content
(as a result of the inability to utilize oxygen). Another early sign of cyanide
toxicity is the acute resistance to the hypotensive effects of increasing doses
of sodium nitroprusside (tachyphylaxis). It should be noted that tachyphylaxis
implies acute tolerance to the drug following multiple rapid injections, as
opposed to tolerance, which is caused by more chronic exposure. Cyanide
toxicity is more likely
if the cumulative dose of sodium
nitroprusside is greater than 500 mcg/kg administered at an infusion rate
faster than 2 mcg/kg/min. Patients with cyanide toxicity should be mechanically
ventilated with 100% oxygen to maximize oxygen availability. The
phar-macological treatment of cyanide toxicity depends on increasing the
kinetics of the two reactions by administering sodium thiosulfate (150 mg/kg
over 15 min) or 3% sodium nitrate (5 mg/kg over 5 min), which oxidizes
hemoglobin to methemoglobin. Hydroxocobalamin combines with cyanide to form
cyanocobalamin (vitamin B12).
Thiocyanate is slowly cleared by the
kidney. Accumulation of large amounts of thiocyanate (eg, in patients with
renal failure) may result in a milder toxic reaction that includes thyroid
dysfunction, muscle weakness, nausea, hypoxia, and an acute toxic psychosis.
The risk of cyanide toxicity is not increased by renal failure, however.
Methemoglo-binemia from excessive doses of sodium nitroprus-side or sodium
nitrate can be treated with methylene blue (1–2 mg/kg of a 1% solution over 5
min), which reduces methemoglobin to hemoglobin.
Effects on Organ Systems
The combined dilation of venous and
arteriolar vascular beds by sodium nitroprusside results in reductions of
preload and afterload. Arterial blood pressure falls due to the decrease in
peripheral vas-cular resistance. Although cardiac output is usu-ally unchanged
in normal patients, the reduction in afterload may increase cardiac output in
patients with congestive heart failure, mitral regurgitation, or aortic
regurgitation. In opposition to any favor-able changes in myocardial oxygen
requirements are reflex-mediated responses to the fall in arterial blood
pressure. These include tachycardia and increased myocardial contractility. In
addition, dilation of cor-onary arterioles by sodium nitroprusside may result
in an intracoronary steal of blood
flow away from ischemic areas that are already maximally dilated.
Sodium nitroprusside dilates cerebral
vessels and abolishes cerebral autoregulation. Cerebral blood flow is
maintained or increases unless arterial blood pressure is markedly reduced. The
resulting increase in cerebral blood volume tends to increase intracranial
pressure, particularly in patients withreduced intracranial compliance (eg,
brain tumors). This intracranial hypertension can be minimized by slow
administration of sodium nitroprusside and institution of hypocapnia.
The pulmonary vasculature also dilates
in response to sodium nitroprusside infusion. Reduc-tions in pulmonary artery
pressure may decrease the perfusion of some normally ventilated alveoli,
increasing physiological dead space. By dilating pulmonary vessels, sodium
nitroprusside may prevent the normal vasoconstrictive responseof the pulmonary
vasculature to hypoxia (hypoxic pulmonary vasoconstriction). Both of these
effects tend to mismatch pulmonary ventilation to perfusion and decrease
arterial oxygenation.
In response to decreased arterial blood
pressure, renin and catecholamines are released during administration of
nitroprusside. Renal function is fairly well maintained during sodium
nitroprusside infusion, despite moderate drops in arterial blood pressure and
renal perfusion.
Sodium nitroprusside does not directly
interact with neuromuscular blocking agents. Nonetheless, a decrease in muscle
blood flow caused by arterial hypotension could indirectly delay the onset and
prolong the duration of neuromuscular blockade.
Related Topics