Chapter: Clinical Dermatology: Other genetic disorders
Neurofibromatosis
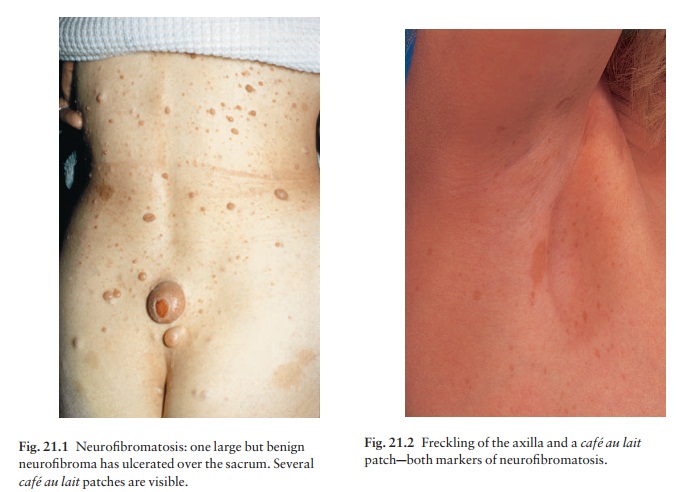
Neurofibromatosis
This
relatively common disorder affects about 1 in 3000 people and is inherited as
an autosomal dominant trait. There are two main types: von RecklinghausenŌĆÖs
neurofibromatosis (NF1; which accounts for 85% of all
cases) and bilateral acoustic neurofibromatosis (NF2); these
are phenotypically and genetically distinct.
Cause
The
NF1
gene has been localized to chromosome 17q11.1. It is unusually large (300 kb)
and many dif-ferent mutations within it have now been identified. The NF1
gene is a tumour suppressor gene, the product of which, neurofibromin,
interacts with the product of the RAS proto-oncogene. This may explain
the sus-ceptibility of NF1 patients to a variety of tumours.
The inheritance of NF1 is as an autosomal dominant trait
but about one-half of index cases have no preced-ing family history.
The
inheritance of NF2
is also autosomal domin-ant. Mapping to chromosome 22q12.2 followed the
observation of changes in chromosome 22 in menin-giomas as these tumours may be
seen in NF2.
This gene also normally functions as a tumour-suppressor gene, the product of
which is known as schwannomin.
Clinical features
The physical signs include the following
Von RecklinghausenŌĆÖs neurofibromatosis (NF1)
ŌĆó
Six or more caf├® au
lait patches (light brown oval macules; Fig. 21.1), usually
developing in the first year of life.
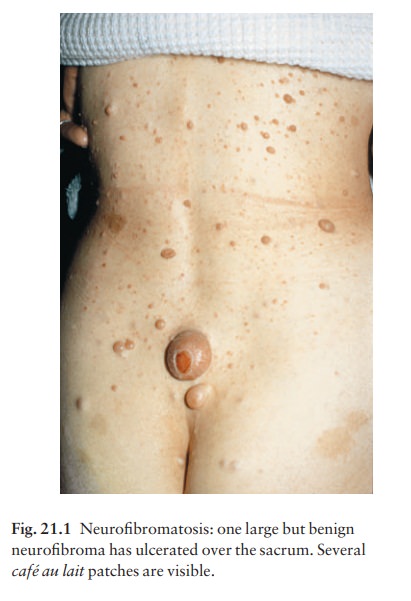
ŌĆó
Axillary freckling (Fig. 21.2) in
two-thirds of affected individuals.
ŌĆó
Variable numbers of skin
neurofibromas, some small and superficial, others larger and deeper, ranging
from flesh-coloured to pink, purple or brown (Fig. 21.1). Most are dome-like
nodules, but others are irregular raised plaques. Some are firm, some soft and
com-pressible through a deficient dermis (ŌĆśbutton-holeŌĆÖ sign); others feel
ŌĆśknottyŌĆÖ or ŌĆśwormyŌĆÖ. Neurofibromas may not appear until puberty and become
larger and more numerous with age.
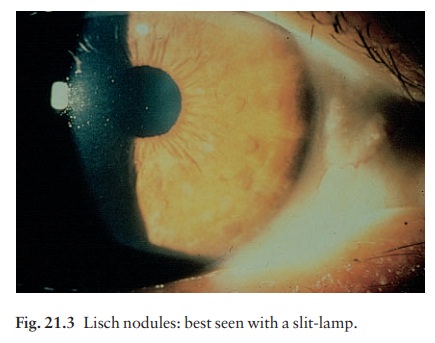
ŌĆó
Small circular pigmented hamartomas
of the iris (Lisch nodules; Fig. 21.3), appear in early childhood.
Nearly all NF1 patients meet the criteria for dia-gnosis by the age of 8 years, and all do so by 20 years. The usual order of appearance of the clinical features is caf├® au lait macules, axillary freckling, Lisch nodules and neurofibromas.
Bilateral acoustic neurofibromatosis (NF2)
ŌĆó
Bilateral acoustic neuromas.
ŌĆó
Few, if any, cutaneous
manifestations.
ŌĆó
No Lisch nodules.
Diagnosis
The
caf├® au
lait marks, axillary freckling and Lisch nodules should be
looked for, as they appear before the skin neurofibromas. A segmental form of
NF1 is caused by a postzygotic mutation. Isolated neurofi-bromas are not
uncommon in individuals without neurofibromatosis and are of little consequence
unless they are painful.
Complications
Von RecklinghausenŌĆÖs neurofibromatosis
A
neurofibroma will occasionally change into a neuro-fibrosarcoma. Other
associated features may include kyphoscoliosis, mental deficiency, epilepsy,
renal artery stenosis and an association with phaeochromocy-toma. Forme fruste
variants occur, e.g. segmental neurofibromatosis.
Bilateral acoustic neurofibromatosis
Other
tumours of the central nervous system may occur, especially meningiomas and
gliomas.
Management
Ugly
or painful lesions, and any suspected of undergo-ing malignant change, should
be removed. The chance of a child of an affected adult developing the disorder
is 1 in 2aparents should be advised about this.
Related Topics