Chapter: Pathology: Red Blood Cell Pathology: Anemias
Microcytic Anemias
MICROCYTIC ANEMIAS
Iron Deficiency Anemia
Iron physiology. Functionally
available iron is normally found in hemoglobin,myoglobin, and enzymes (catalase
and cytochromes). Additionally, ferritin is the physiological storage form
(plasma ferritin is normally close to the total body Fe), and hemosiderin
(Prussian blue positive) is iron precipitated in tissues in the form of
degraded ferritin mixed with lysosomal debris.
Iron
is transported in the blood stream by transferrin. Transferrin saturation is
reported as a percentage; it represents the ratio of the serum iron to the
total iron-binding capacity, multiplied by 100.
Dietary
deficiency of iron is seen in elderly populations, children, and the poor.
Increased demand for iron is seen in children and pregnant women. Additionally,
iron deficiency can develop because of decreased absorption, either due to
general-ized malabsorption or more specifically after gastrectomy (due to
decreased acid, which is needed for ferrous absorption) or when there is
decreased small intesti-nal transit time (causing “dumping syndrome”). Iron
deficiency can also be due to chronic blood loss due to gynecologic (menstrual
bleeding) or gastrointestinal causes (in the United States, think carcinoma; in
the rest of the world, think hookworm).
The
sequence of events during iron deficiency is as follows:
·
Initially,
decreased storage iron results in decreased serum ferritin anddecreased bone marrow
iron on Prussian blue stains.
·
The next stage is decreased circulating iron, which causes decreased
serum iron, increased total iron binding capacity, and decreased % saturation.
·
The last stage is formation of microcytic/hypochromic anemia, with decreased
MCV, decreased MCHC, and high RDW.
Other clinical features of iron
deficiency include increased free erythrocyte protoporphyrin (FEP),
oral epithelial atrophy if Plummer-Vinson syndrome is present, koilonychia
(concave or spoon nails with abnormal ridging and splitting), and pica (eating
nonfood substances, e.g. dirt).
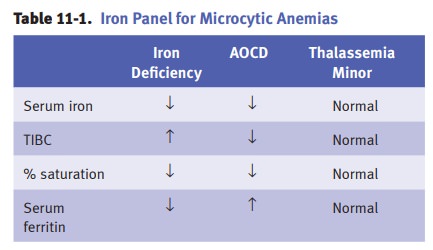
Anemia of chronic disease (AOCD)
(or anemia of inflammation) is characterizedby iron being trapped in bone
marrow macrophages, leading to decreased utilization of endogenous iron stores.
Laboratory studies show increased serum ferritin with decreased total iron
binding capacity. Increased IL-6 increases plasma hepcidin, which is a negative
regulator of iron uptake in the small intestine and of iron release from
macrophages.
Thalassemia
syndromes are quantitative, not qualitative, abnormalities of
hemo-globin. α-thalassemia
has decreased α-globin
chains with relative excess β
chains, while β-thalassemia
has decreased β-globin
chains with relative excess α
chains. It is hypothesized that the thalassemia genes have been selectively
preserved in the human genome because the thalassemias provide a protective
advantage to carriers exposed to diseases such as malaria.
α-thalassemia.There are a total of 4 α- globin chain genes, 2 from each
parent.α-thalassemia
is due to gene deletions in the α-globin
chain genes, and the clini-cal manifestations depend upon the number of genes
that are affected. α chains
are normally expressed prenatally and postnatally; therefore, there is prenatal
and postnatal disease. In normal individuals, 4 α
genes (αα/αα) are present and 100% of the α chains are normal.
·
In the silent carrier state, one deletion is present, and the total number
ofgenes available is 3 (– α/αα), which produce 75% of the needed α chains. Individuals with the silent carrier state are
completely asymptomatic and all lab tests are normal.
·
In α-thalassemia trait,
2 deletions are present, and the total number of avail-able α genes is 2, which produce 50% of the needed α chains. The genotype cis (– –/αα)
is seen in Asians, while the genotype trans (–α/–α) is seen in African Americans (offspring don’t develop
hemoglobin H disease or hydrops fetalis).
·
Hemoglobin
H disease is characterized by 3 deletions, with the number ofαgenes being 1 (– –/– α),
which produces 25% of the normal α
chains. There is increased Hb H (β4,)
which forms Heinz bodies that can be seen with crystal blue stain.
·
Hydrops fetalis has 4 deletions and is lethal in
utero, because the number ofgenes is 0 (– –/– –), producing 0% α chains.
β-thalassemia.There are a total of 2 β-globin chain genes. In contrast to
the
α-globinchain
genes, the 2 β-globin
chain genes are expressed postnatally only, and therefore there is only
postnatal disease and not prenatal disease. The damage to the genes is mainly
by point mutations, which form either some β chains (β+) or none (β0).
·
β-thalassemia
minoris
seen when one of the β-globin chain genes has beendamaged. The condition is
asymptomatic, and characterized on laboratory studies by increased hemoglobin
A2 (8%) and increased hemoglobin F (5%).
·
β-thalassemia intermediacauses
varying degrees of anemia, but no transfu-sions are needed.
·
β-thalassemia major(Cooley
anemia). Patients are normal at birth, and symp-toms develop at about 6 months
as hemoglobin F levels decline. Severe hemo-lytic anemia results from decreased
erythrocyte life span. This severe anemia causes multiple problems:
§
Intramedullary destruction results
in “ineffective erythropoiesis.”
§
Hemolysis causes jaundice and an
increased risk of pigment (bilirubin) gallstones.
§
Lifelong transfusions are required,
which result in secondary hemo chromatosis.
§
Congestive heart failure (CHF) is
the most common cause of death.
Erythroid
hyperplasia in the bone marrow causes “crewcut” skull x-ray and increased size
of maxilla (“chipmunk face”). The peripheral blood shows microcytic/hypo-chromic
anemia with numerous target cells and increased reticulocytes. Hemo-globin
electrophoresis shows increased hemoglobin F (90%), normal or increased
hemoglobin A2, and decreased hemoglobin A. Treatment is hematopoietic stem cell
transplantation.
Sideroblastic
anemia is a disorder in which the body has adequate iron stores,
but isunable to incorporate the iron into hemoglobin. It is associated with
ring sideroblasts (accumulated iron in mitochondria of erythroblasts) in bone
marrow. Sideroblastic anemia may be either pyridoxine (vitamin B6) responsive
or pyridoxine unrespon-sive; the latter is a form of myelodysplastic syndrome
(refractory anemia with ring sideroblasts). The peripheral blood may show a
dimorphic erythrocyte population. Laboratory studies show increased serum iron,
ferritin, FEP, and % saturation of TIBC, with decreased TIBC.
Related Topics