Chapter: Essential Clinical Immunology: Immunological Aspects of Chest Diseases: The Case of Tuberculosis
Host-Pathogen Interactions in TB
Host-Pathogen Interactions in TB
Upon entering the respiratory tract, bacilli reaching the lung alveoli are phagocytosed by resident macrophages through man-nose, complement, and Fc receptors (in differentiated macrophages). As dem-onstrated more than three decades ago by Armstrong and Hart (1975), bacterial uptake through Fc receptors increases phagolysosomal fusion, triggers the oxida-tive burst, and leads to bacterial killing in human macrophages ex vivo. Regardless of the receptor involved, uptake apparently requires recruitment of cholesterol to the nascent phagosome. Uptake by resident DCs is believed to occur through DC-SIGN adhesion.
CELLULAR INTERACTIONS LEADING TO A PROTECTIVE IMMUNE RESPONSE
A protective immune reponse to M. tuberculosis involves mainly the T-cell-mediated branch of adaptive immunity.
As early as one week postinfection, bacte-ria-specific CD4+ and CD8+ T cells migrate to the lung, where they peak in numbers at around four weeks postinfection, Studies in immune-deficient mice have established that IFN-γ, TNF-α, and IL-12 are the main cytokines involved in orchestrating a pro-tective immune response after M. tubercu-losis infection. The relevance of IFN-γ and IL-12 in antimycobacterial immunity is sup-ported by human genetic data; the recent association between administration of TNF-α inhibitors used to treat rheumatoid arthri-tis and the reactivation of LTBI has under-scored the protective role of this cytokine in control of M. tuberculosis in humans.
The initiation of the immune response occurs through presentation of M. tuber-culosis antigens, presumably by DCs, on MHC class I and II molecules to naïve T cells, which in the context of IL-12 and IL-23 produced by infected macrophages and DCs themselves, are instructed to dif-ferentiate into IFN-γ-secreting cells (helper T type 1, TH1) cells (see Figure 1). Both CD4+ and CD8+ T cells contribute to the activation of infected macrophages through IFN-γ secretion, while CD8+ T cells might additionally contribute to protection by causing apoptosis of infected macrophages through the Fas-FasL pathway, as well as through secretion of granulysin and other microbicidal molecules. However, the relative contribution of CD8+ T cells to the overall protective cell-mediated immunity might not be as significant as that by CD4+ T cells because M. tuberculosis–infected mice deficient in the former live almost as long as infected wild-type mice, while those deficient in the latter are significantly more susceptible.
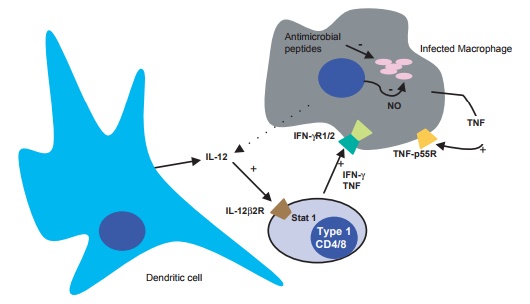
Figure 13.1 Cellular immune responses to M. tuberculosis infection. Some aspects of the innate and adaptive cellular immune responses to infection by M. tuberculosis are shown as inferred from studies using ex vivo and in vivo models of tuberculosis. Bacteria are assumed to reside mostly inside vacuoles in macrophages. Not shown are CTL- and gamma-, delta-T-cell-mediated proposed mechanisms of protection.
Mice lacking CD4+ T cells exhibit reduced macrophage activation, which is associated with lower IFN-γ production, and are therefore highly susceptible to M. tuberculosis infection. Even though CD8+ T-cell production of IFN-γ can com-pensate and restore the concentrations of IFN-γ back to normal levels in CD4+ T-deficient mice, the latter still succumb to the infection earlier than wild-type mice, suggesting that CD4+ T-cell-derived IFN-γ production early in the infection, or an IFN-γ-independent mechanism might be important for control of the infection. An IFN-γ-independent CD4+ T-cell-mediated mechanism dependent on NOS2 has recently been shown in vitro and sug-gested to be important in vivo by the dem-onstration that IFN-γ-deficient mice can be effectively protected by BCG vaccination in a CD4+ T-cell-dependent manner.
Cell-mediated immunity to M. tubercu-losis promotes the formation of granulo-mas – infected macrophages surrounded by antigen-specific lymphocytes, mul-tinucleated giant cells, and a ring of fibrous deposition. Granulomas typically encompass the entire infection foci and effectively isolate the bacilli from the rest of the lung. Granuloma formation depends largely on TNF-α and GM-CSF, which are produced by infected macrophages, T cells, and other hemaetopoetic cells, and which drive tissue-resident stromal cells to produce macrophage-inflamma-tory protein-1β (MIP-1β), MIP-1α, and other chemokines needed to recruit neu-trophils, lymphocytes, and monocytes to the site of infection. TNF-α-/- mice infected with M. tuberculosis fail to form organized granulomas, are incapable of controlling M. tuberculosis replication, and succumb to M. tuberculosis within 20 to 30 days of infection. Importantly, the levels of TNF-α must be tightly regulated because overpro-duction can lead to excessive pathology.
SURVIVAL OF M. TUBERCULOSIS IN THE MACROPHAGE
Pathogenic mycobacteria reside and readily replicate within tight vacuoles of non-immune-activated macrophages. Pio-neering studies by Rees and Hart (1961) showed that tubercle bacilli prevent the fusion of lysosomes to M. tuberculosis– containing vacuoles, thus preventing the vacuoles’ normal acidification and facili-tating bacterial survival and replication. Vacuoles containing live virulent Mycobac-terium avium have a relatively high pH (6.3– 6.5), which correlates with a lower density of vesicular H+ ATPase pumps on their membranes. Mycobacterial vacuoles reside within the recycling and sorting endosomal compartment – markers of early endo-somes (Rab5) are retained, markers of late endosomes (Rab7) and lysosomes (LAMP1-1 and LAMP-2) are largely excluded; communication with the cytoplasmic mem brane appears to be maintained, as suggested by the cycling of transferrin in and out of the vacuoles. Diversion of the mycobacterial phagosome from the normal maturation pathway correlates with reten-tion of the actin-binding coronin TACO on the phagosomal membrane. Active evasion of phagolysosomal fusion by M. tuberculosis involves bacterial effectors such mannose-capped lipoarabinomannan (ManLAM), which interferes with phagolysosomal fusion by blocking the acquisition of the lysosomal cargo and syntaxin 6 from the trans-Golgi network. LAM alone can spe-cifically block the cytosolic Ca2+ increase necessary for a signaling cascade involving phosphatidylinositol (PI) 3 kinase hVPS34, which is needed for production of PI 3 phosphate (PI3P) on phagosomes.
The importance of type 1 cytokines in human immunity to M. tuberculosis infec-tion has been confirmed by studies of naturally occurring polymorphisms in the genes encoding type 1 cytokines and their receptors in humans. The principal role of type 1 cytokines in anti-TB immunity is to activate the antimicrobial functions of mac-rophages. IFN-γ activation and infection of murine macrophages with M. tuberculo-sis results in the differential expression of some 40 percent of the macrophage genes, an indication of just how dynamic the host–M. tuberculosis interaction is. Many of these differentially expressed genes likely encode functions involved in the curtailment of growth and killing of the intracellular bacilli.
EFFECTOR MECHANISMS OF THE IMMUNE-ACTIVATED MACROPHAGE
Schaible and colleagues (1998) found that upon activation of M. avium–infected macrophages with IFN-γ and lipopolysac-charide (LPS), the mycobacterial vacuoles fuse to each other, become more acidic (pH 5.3), and as a result of fusion with lyso-somes accumulate hydrolases. These and other changes in the intracellular milieu lead to stasis and killing of the bacilli.
MacMicking and colleagues (2003) indicate that LRG47, a member of a family of IFN-γ–dependent GTPases that mediate fusion events in endosomal networks, is one of the facilitators of phagolysosomal fusion and acidification downstream of IFN-γ and is essential for protection. These GTPases play different functions in response to infection by different pathogens as sug-gested by the findings that IGTP–/– and IRG-47–/– mice, which are susceptible to infection by viral, bacterial, and eukaryotic pathogens are resistant to infection by M. tuberculosis. In contrast, LRG47-/- mice suc-cumb to infection by M. tuberculosis with kinetics similar to those of NOS2-/- mice, the other major pathway of antimycobacte-rial immunity.
Work from the laboratories of Carl Nathan and Ferric Fang over the years has demonstrated that the immune system relies on chemically reactive micromol-ecules to neutralize and eliminate invading pathogens and their products. Some of the most efficient of these reactive molecules in killing and controlling M. tuberculosis include the reactive nitrogen intermediates derived from nitric oxide (NO) produced by NO synthase 2. M. tuberculosis is killed by murine peritoneal macrophages and cell lines activated with IFN-γ and either LPS or TNF-α through reactive nitrogen inter-mediate (RNI) production, and IFN-γ and TNF-α synergistically increase NO synthe-sis by macrophages. The demonstration that NOS2-/- mice are highly susceptible to M. tuberculosis infection provides clear functional genetic evidence that NOS2 is essential for the protective immune response against M. tuberculosis.
Even though macrophage activation and resulting RNI production result in bac-teriostasis, M. tuberculosis is nonetheless able to resist eradication in mouse lungs and in activated macrophages in tissue cul-ture, despite the fact that in vitro reagent NO is a potent mycobactericidal. Having evolved to infect the mammalian lung, M. tuberculosis appearently has ways of countering the nossive effects of NO and related RNI. One such mechanism involves the product of the mpa gene, which forms hexamers that have ATPase activity, and is believed to be associated with a myco-bacterial proteasome. mpa M. tuberculosis bacteria are attenuated in wild-type mice and show some reversion to virulence in NOS2-/-. It remains to be shown whether mpa M. tuberculosis bacteria have defects in protein degradation through this presumptive proteasome, and whether this proposed function is involved in protect-ing the bacteria from RNI. An additional strategy adopted by M. tuberculosis to deal with RNI appears to be simple avoidance of NOS2. Recent microscopy studies indi-cate that M. tuberculosis might be able to prevent NOS2 from being recruited to bacteria-containing phagosomes.
The role of NO in protection against M. tuberculosis in humans is somewhat con-troversial. While some investigators have reported NOS2 protein induction, NO, which is measured by determining the concentration of its breakdown products nitrate (NO3) and nitrite (NO2), cannot be consistently detected in M. tuberculosis– infected cultures of human monocytes/ macrophages. Furthermore, no mutations in nos2 in humans have been associated with susceptibility to M. tuberculosis. How-ever, patients with active TB apparently exhale high levels of NO, which correlate with high expression of NOS2 and spon-taneous production of nitrite by alveolar macrophages.
Another reactive micromolecule that is a potent antimicrobial effector of the immune response is the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (PHOX)-mediated production of reactive oxygen intermediates (ROIs) including reduction products of O2 such as superoxide, hydrogen peroxide, and hydroxyl radical. ROI generation is an effec-tive antimicrobial defense against many bacterial pathogens including Salmonella and Burkholderia cepacia. However, recent work from John McKinney’s laboratory indicates that the protective role of ROI in TB appears to be less prominent than dur-ing Salmonella infections. Chronic granu-lomatous disease (CGD) patients that lack phagocyte oxidase have not been reported to be more susceptible to M. tuberculosis infection. Furthermore, gp91(phox-/-) mutant mice, which lack a functional phago-cyte oxidase, are not more susceptible to M. tuberculosis than wild-type mice, while mice lacking the gp47 subunit of PHOX are less able to control M. tuberculosis replication only transiently. Furthermore, M. tuberculosis appears to be able to effec-tively counteract the damaging effects of ROIs. ROI detoxification mediated by the KatG catalase is essential for bacterial per-sistence in mice following induction of adaptive immunity, and an M. tuberculosis mutant of katG is attenuated in wild-type mice, but reverts to wild-type levels of vir-ulence in mice lacking PHOX. The product of katG is also a peroxynitritetase, thus it may protect M. tuberculosis from both RNI and ROI. At least one of the superoxide dismutases of M. tuberculosis contributes to intracellular survival.
IMMUNITY DURING M. TUBERCULOSIS CHRONIC INFECTION
Several of the components that are important for generating a protective immune response to M. tuberculosis infec-tion are also important for maintaining the equilibrium between host and pathogen during the chronic phase of the infection. As expected, TH1 maintenance is important for controlling M. tuberculosis during the chronic phase of the infection, and sustain-ing this type of response requires IL-12. Neutralization of TNF-α by antibodies or by infection with an adenovirus express-ing a soluble TNF-α receptor resulted in marked increases in bacterial loads and exacerbation of disease in the mouse model of chronic TB and in a variation of the Cor-nell model of drug-induced latency. Work from the laboratories of John Chan and Joan Flynn using the Cornell model of TB, has demonstrated that IFN-γ is involved in preventing reactivation. Similarly, chemical inhibition of NOS2 in chronically infected mice resulted in rapid increases in lung bacterial loads. NOS2-inhibiton also reactivated M. tuberculosis in variations of the Cornell model of latency.
EXTRACELLULAR PERSISTENCE OF M. TUBERCULOSIS IN VIVO
In the human lung, M. tuberculosis is thought to reside both intracellularly and extracellularly, depending on the type of lesion. Pioneering work by Lurie (1964) and continued by Dannenberg using the rabbit model showed the existence of extracellular M. tuberculosis in caseous necrotic lesions. Because immunity to TB was thought to be mainly mediated by acti-vated phagocytes that kill intracellular bac-teria, it was suspected that the extracellular milieu represented a safe heaven for bacte-rial replication and persistence. In order to study this population of bacteria directly back in 1939, Lurie devised a very inge-nious method of cultivating bacteria extra-cellularly in vivo: Parlodion-film–encased silk bags that were impervious to host cells, but not to plasma, were inoculated with bacilli and then implanted subcutane-ously in rabbits. These extracellular bacte-ria replicated unhindered during the first two weeks of infection, but then leveled off at around two weeks postinfection. When immunized animals were studied, bacteria stopped growing earlier and were killed more efficiently than in naïve counterparts, strongly implicating acquired immunity in controlling the growth of extracellular bac-teria. Because control of growth correlated with a more rapid mobilization of mono-nuclear cells toward the encased focus of infection, it was proposed that factors secreted by activated macrophages ren-dered the fluids entering the infection foci inimical to bacterial replication.
Recently, work in William Bishai’s laboratory adapted Lurie’s strategy to the widely used model of murine TB: M. tubercu-losis was encapsulated in cell-impermeable hollow fibers, and these were implanted subcutaneously into mice for in vivo cultivation. As expected, an initial period of bacterial replication was followed by cur-tailment of growth, followed by a consider-able decline in numbers, and, interestingly, an eventual stabilization of the counts. These stationary-state colony forming unit (CFU) counts, similarly to those in the equi-librium that characterized the chronic phase of infection in the mouse model of chronic TB as described by Rees and Hart in 1965, represented a somewhat static scenario: they found that about half of the encapsu-lated bacilli were alive by differential stain-ing; a highly dynamic equilibrium would have resulted in a large accumulation of dead bacteria and a decreasing proportion of live ones. Furthermore, indirect analysis – comparison of ATP utilization and CFU at corresponding time points – indicated that soon after implantation, bacteria decreased their metabolism. The rapid cessation of multiplication and rather swift reduction in metabolic activity suggest that innate defenses or perhaps immune-independent mechanisms prompt these adaptations. These adaptations were also suggested by Lurie’s studies using vaccinated animals, which implicated an unknown plasma fac-tor in bacterial growth inhibition. This fac-tor could be nitric oxide, which is known to block M. tuberculosis replication in vitro by inhibiting bacterial respiration. Care-ful analysis of the hollow fiber’s contents in this extracellular model of persistence in vivo, including concentrations of NO, oxygen, essential elements, and nutrients, might be of value.
Related Topics