Chapter: Pathology: Circulatory Pathology
Hemostasis and Bleeding Disorders
HEMOSTASIS AND BLEEDING DISORDERS
Hemostasis is
a sequence of events leading to the cessation of bleeding by the forma-tion of
a stable fibrin-platelet hemostatic plug. It involves interactions between the
vascular wall, platelets, and the coagulation system.
Vascular Wall Injury
Transient
vasoconstriction is mediated by endothelin-1. Thrombogenic factors include a
variety of processes:
·
Changes in blood flow cause turbulence and stasis favor clot
formation.
·
Release of tissue factor from injured cells activates factor
VII (extrinsic pathway).
·
Exposure of thrombogenic subendothelial collagen activates
factor XII (intrin-sic pathway).
·
Release of von Willebrand factor (vWF) binds to exposed
collagen and facili-tates platelet adhesion.
·
Decreased endothelial synthesis of antithrombogenic substances
(prostacy-clin, nitric oxide [NO2], tissue plasminogin activator,
and thrombomodulin)
Platelets
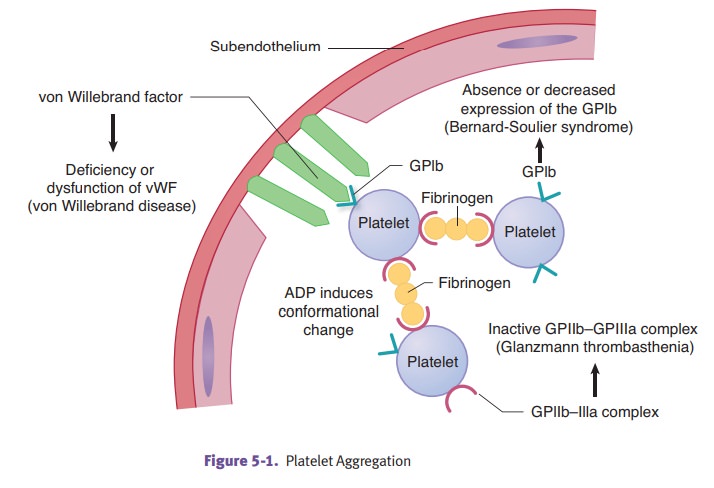
Platelets are
derived from megakaryocytes in the bone marrow. They form a throm-bus through a
series of steps.
·
Step 1: Platelet adhesion occurs when vWF adheres to
subendothelial collagenand then platelets adhere to vWF by glycoprotein Ib.
·
Step 2: Platelet activation occurs when platelets undergo a
shape change anddegranulation occurs. Platelets synthesize thromboxane A2.
Platelets also show membrane expression of the phospholipid complex, which is
an impor-tant substrate for the coagulation cascade.
·
Step 3: Platelet aggregation occurs when additional platelets are
recruited fromthe bloodstream. ADP and thromboxane A2 are potent mediators of
aggrega-tion. Platelets bind to each other by binding to fibrinogen using
GPIIb-IIIa.
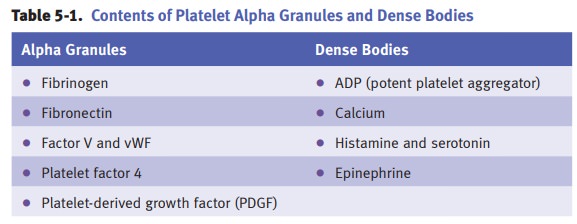
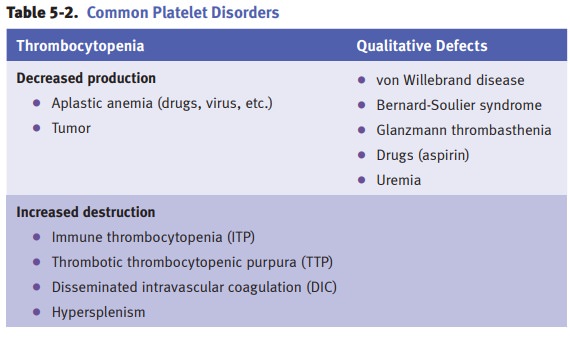
Laboratory tests for platelets
include platelet count (normal 150,000–400,000 mm3) and platelet
aggregometry.
Bernard-Soulier
syndrome and Glanzmann thrombasthenia present as mucocutane-ous bleeding in
childhood.
Immune
thrombocytopenia purpura (ITP) is an immune-mediated attack
(usuallyIgG antiplatelet antibodies) against platelets leading to decreased
platelets (throm-bocytopenia) which result in petechiae, purpura (bruises), and
a bleeding diathesis (e.g., hematomas).
The
etiology involves antiplatelet antibodies against platelet antigens such as
GPIIb-IIIa and GPIb-IX (type II hypersensitivity reaction). The antibodies are
made in the spleen, and the platelets are destroyed peripherally in the spleen
by macrophages, which have Fc receptors that bind IgG-coated platelets.
Forms
of ITP include:
·
Acute ITP, seen in children
following a viral infection and is a self-limited disorder.
·
Chronic ITP, usually seen in women
in their childbearing years and may be the first manifestation of systemic
lupus erythematosus (SLE). Clinically, it is characterized by petechiae,
ecchymoses, menorrhagia, and nosebleeds.
Lab
studies usually show decreased platelet count and prolonged bleeding time but
normal prothrombin time and partial thromboplastin time. Peripheral blood smear
shows thrombocytopenia with enlarged immature platelets (megathrombocytes). Bone
marrow biopsy shows increased numbers of megakaryocytes with immature forms.
Treatment
is corticosteroids, which decrease antibody production; immunoglobulin therapy,
which floods Fc receptors on splenic macrophages; and/or splenectomy, which
removes the site of platelet destruction and antibody production.
Thrombotic
thrombocytopenic purpura (TTP) is a rare disorder of hemostasis
inwhich there is widespread intravascular formation of fibrin-platelet thrombi.
It is sometimes associated with an acquired or inherited deficiency of the
enzyme ADAMTS13, responsible for cleaving large multimers of von Willebrand
factor.
Clinically,
TTP most often affects adult women. The inclusion criteria are
microan-giopathic hemolytic anemia and thrombocytopenia, with or without renal
failure or neurologic abnormalities. Pathology includes widespread formation of
platelet thrombi with fibrin (hyaline thrombi) leading to intravascular
hemolysis (throm-botic microangiopathy).
Lab
studies typically show decreased platelet count and prolonged bleeding time but
normal prothrombin time and partial thromboplastin time. Peripheral blood smear
shows thrombocytopenia, schistocytes, and reticulocytosis. Treatment is plasma
exchange.
Hemolytic
uremic syndrome (HUS) is a form of thrombotic
microangiopathy due toendothelial cell damage. It occurs mostly in children,
typically after a gastroenteritis (typically due to Shiga toxin-producing E. coli 0157:H7).
Typical
HUS presents with abdominal pain, diarrhea (an atypical variant is diar-rhea-negative),
microangiopathic hemolytic anemia, thrombocytopenia, and renal failure. Renal
involvement is seen more commonly than in TTP. The kidney shows fibrin thrombi
in the glomeruli. Renal glomerular endothelial cells are targeted by the
bacterial toxin. Glomerular scarring may ensue.
Treatment
is supportive (fluid management, dialysis, erythrocyte transfusions); plasma
exchange is only used for atypical cases.
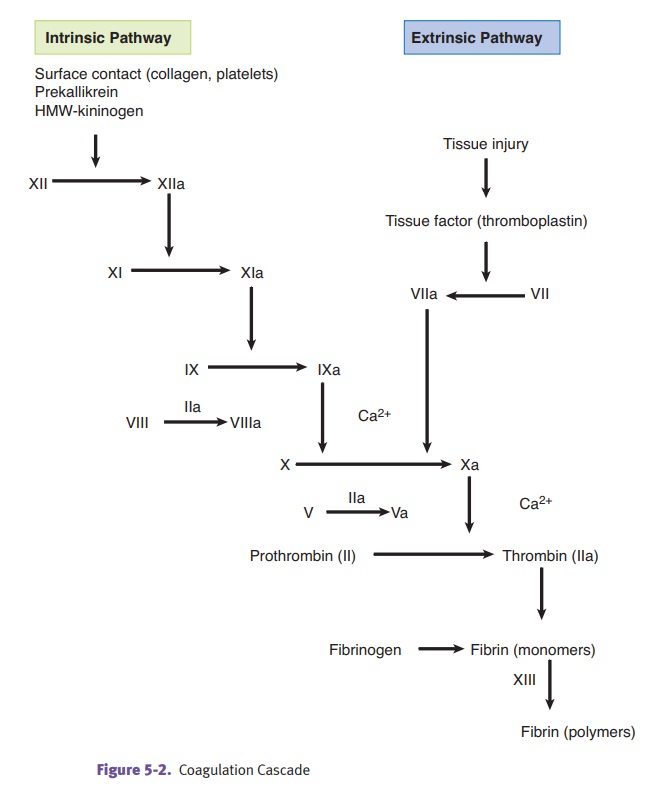
Coagulation factors.The
majority of the clotting factors are produced by the liver.The factors are
proenzymes that must be converted to the active form. Some con- versions occur
on a phospholipid surface, and some conversions require calcium.
·
The intrinsic coagulation pathway is activated by the contact factors,
which include contact with subendothelial collagen, high molecular weight
kinino-gen (HMWK), and kallikrein.
·
The extrinsic coagulation pathway is activated by the release of tissue
factor.
Lab
tests for coagulation include the following:
·
Prothrombin time (PT), which tests the extrinsic and common
coagulation pathways (more specifically, it tests factors VII, X, V,
prothrombin, and fibrin-ogen). The international normalized ratio (INR)
standardizes the PT test so that results throughout the world can be compared.
A longer time means blood takes longer to clot.
·
Partial thromboplastin time (PTT),
which tests the intrinsic and common coagulation pathways (more specifically,
it tests factors XII, XI, IX, VIII, X, V, prothrombin, and fibrinogen).
·
Thrombin time (TT), which tests for
adequate fibrinogen levels.
·
Fibrin degradation products (FDP),
which tests the fibrinolytic system (increased with DIC).
Hemophilia
A (classic hemophilia) is an X-linked recessive condition
resulting froma deficiency of factor VIII. Clinically, hemophilia A
predominately affects males. Symptoms vary depending on the degree of
deficiency.
·
Newborns may develop bleeding at the
time of circumcision.
·
Other problems include spontaneous
hemorrhage into joints (hemarthrosis), easy bruising and hematoma formation
after minor trauma, and severe pro-longed bleeding after surgery or
lacerations.
Laboratory
studies typically show normal platelet count and normal bleeding time, normal
PT and prolonged PTT. Treatment is factor VIII concentrate.
Hemophilia
B (Christmas disease) is an X-linked recessive condition
resulting froma deficiency of factor IX that is clinically identical to
hemophilia A. Treatment is recombinant factor IX.
Acquired
coagulopathies include vitamin K deficiency (decreased synthesis of
fac-tors II, VII, IX, X, and protein C & S) and liver disease (decreased
synthesis of virtually all clotting factors).
Von
Willebrand disease is an autosomal dominant bleeding
disorder characterizedby a deficiency or qualitative defect in von Willebrand
factor. vWF is normally produced by endothelial cells and megakaryocytes.
Clinical features include spon-taneous bleeding from mucous membranes,
prolonged bleeding from wounds, and menorrhagia in young females. Hemarthrosis
is uncommon.
Lab
studies show normal platelet count, a prolonged bleeding time, normal PT, and
often prolonged PTT. Abnormal platelet response to ristocetin (adhesion defect)
is an important diagnostic test. Treatment for mild classic cases (type I) is
desmo-pressin (an antidiuretic hormone analog), which releases vWF from
Weibel-Palade bodies of endothelial cells.
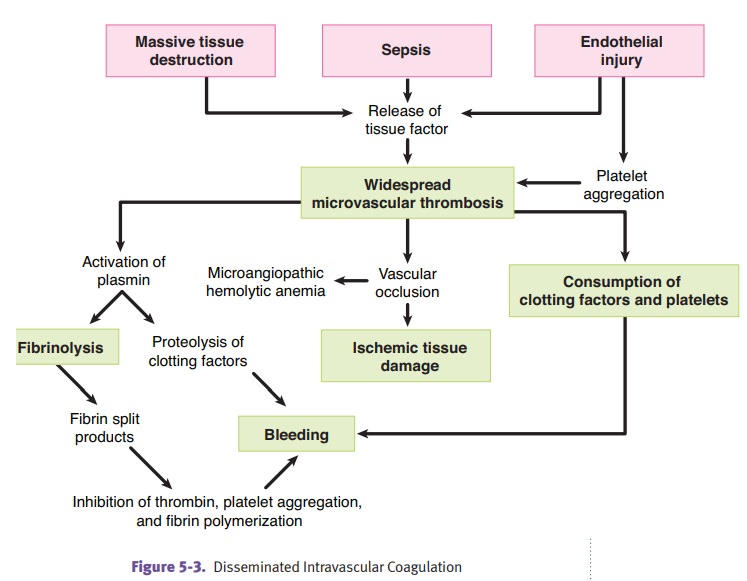
Disseminated
intravascular coagulation (DIC) is always secondary to another
dis-order. Causes are diverse.
·
Obstetric complications can cause
DIC because placental tissue factor activates clotting.
·
Gram-negative sepsis can cause DIC
because tumor necrosis factor activates clotting.
·
Microorganisms (especially
meningococcus and rickettsiae)
·
AML M3 (cytoplasmic granules in
neoplastic promyelocytes activate clotting)
·
Adenocarcinomas (mucin activates
clotting)
DIC
causes widespread microthrombi with consumption of platelets and clotting
factors, causing hemorrhage. Laboratory studies show decreased platelet count,
pro-longed PT/PTT, decreased fibrinogen, and elevated fibrin split products (D
dimers). Treat the underlying disorder.
Related Topics