Chapter: Essential Clinical Immunology: Immunological Aspects of Transplantation
Hematopoietic Stem Cell Transplantation
HEMATOPOIETIC STEM CELL TRANSPLANTATION
HSCT involves the infusion of immature blood-forming cells into the circulation of a patient to reconstitute the recipient’s bone marrow. HSCT is a promising form of therapy for patients with certain forms of genetic diseases, specific cancers, and vari-ous blood disorders (Table 18.1). The trans-planted hematopoietic stem cells (HSCs) may come from the patient (autologous HSCT) or from a different stem cell donor (allogeneic HSCT).
The field of clinical HSCT began in the late 1940s, when experiments in mice showed that shielding of the spleen allowed animals to survive oth-erwise lethal total-body irradiation. It was not until the seminal publication by E. Donnall Thomas in the 1957 NewEngland Journal of Medicine, however, that even transiently successful hema-topoietic transplants were first reported in humans; and at least another decade passed before allogeneic marrow graft-ing really began to have clinical suc-cesses. Thomas was later recognized with the 1990 Nobel Prize in Medicine for his pioneering work in HSCT. Outcomes continued to improve through the 1970s as HLA matching became commonplace, allowing expansion of the clinical indi-cations for transplant to patients who did not have end-stage disease and who were in better clinical condition. Fur-ther expansion of transplant availabil-ity between nonsibling donor-recipient pairs has driven refinements in HLA typ-ing, allowing better matching with high-resolution, DNA-based methods, rather than serologic methods, at the allelic subtype level for class I and II MHC antigens.
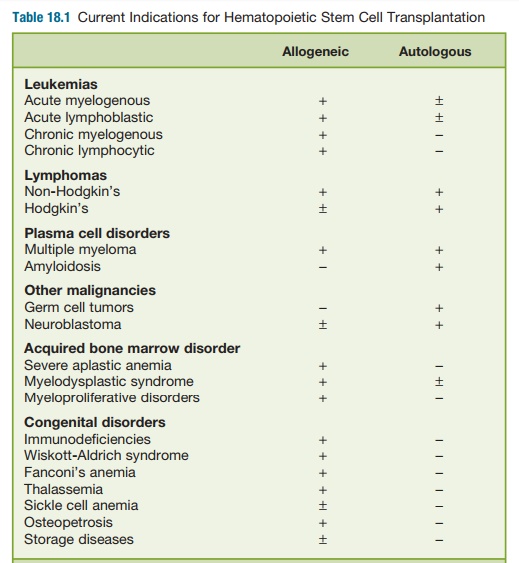
Hematopoietic Stem Cell Transplant Sources
The stem cell product was originally harvested directly from bone marrow under general anesthesia and used almost exclusively in the allogeneic setting. With the advent of the cytokine era, G-CSF given during recovery from treatment with cyclophosphamide, which spares the actual CD34+ HSCs, has proven effec-tive in mobilizing CD34+ HSCs into the circulation for collection by leukopheresis. These peripheral blood stem cells (PBSC) are now in routine use for autologous transplantation.
G-CSF-mobilized PBSCs, without prior cyclophosphamide treatment, have also largely replaced steady state bone marrow harvesting in allogeneic transplantation. Randomized comparison trial between these two stem cell sources are ongo-ing. The concentration of CD34+ HSCs in normal resting bone marrow is up to 100 times higher than in peripheral blood. A daily dose of at least 10 µg/kg of subcu-taneously administered G-CSF (filgrastim) for four to six days increases the num-ber of CD34+ cells in the blood fiftyfold or higher, or to levels comparable to or greater than those in marrow. One or two peripheral blood leukopheresis collections from G-CSF-primed healthy donors on consecutive days will result in a CD34+ cell collection that is sufficient for the most allogeneic transplants. In the short term, this mobilization regimen is benign, although 80 percent of recipients will develop bone pain and 50 percent develop headaches due to the increased cellularity and turnover of marrow in its closed space (the skull also contains marrow). To date, there have been no long-term safety issues.
CD34+ HSCs and their absolute number per kilogram of recipient weight are criti-cal determinants of eventual engraftment, regardless of the stem cell source.
Hematopoietic recovery occurs earlier after transplantation of PBSC than bone mar-row, even though long-term engraftment may be similar. This seems due not only to the dose of CD34+ HSCs but also to other lymphocyte subpopulations included in the PBSC graft. An additional advantage of PBSCs, at least in autografts, is the lower risk that PBSCs are contaminated with tumor cells in the bone marrow.
Finally, umbilical cord blood (UCB) is also an enriched source of CD34+ HSCs that is often otherwise discarded if not cryopreserved for future transplanta-tion use. Shelf life is thought to be at least five years, though viable recoveries vary greatly between different cord blood banks. Cell dose has been a major limi-tation, especially for adults. Pioneering work by Wagner, Barker, and colleagues in this field, however, has established the use of double UCBs to increase cell dose in adults. Interestingly, only one cord establishes long-term engraftment, while the other is lost; and the cord with the larger cell dose does not necessarily per-sist. Engraftment is much slower than with either PBSC or bone marrow allografts, with particularly notable delays in plate-let recovery. Hematopoietic recovery and immunologic recovery are eventually complete and quite robust. Understanding the double-cord biology is an active field of investigation.
Pretransplant Conditioning Regimens
A patient must undergo a conditioning regimen before receiving the HSCT. These regimens fall along a spectrum from fully myeloablative to nonmyeloablative, with reduced intensity regimens in between, and use radiation and/or chemotherapy. These regimens address the two goals of conditioning, which are to provide intensive therapy against the malignancy for which the patient is being treated in the first place and to provide adequate immune suppression to ensure HSC engraftment.
The intensity of the regimen used depends in large part on the aggressiveness of the disease, the remission status, and the degree of HLA matching between donor and recipient (Figure 18.2). Acute leuke-mias and some high-grade, aggressive lym-phomas benefit from fully myeloablative conditioning, which ensures engraftment and protects against recurrent disease. More indolent cancers like low-grade lym-phomas rely heavily on the immunologic activity mediated by the allograft against the malignancy (see sections on graft-versus-leukemia/lymphoma, GvL, or GvT) and require only enough pretransplant conditioning to ensure engraftment. Such reduced intensity and nonmyeloablative regimens also extend allogeneic transplant options to individuals who could not with-stand the toxicities and side effects of fully ablative conditioning.
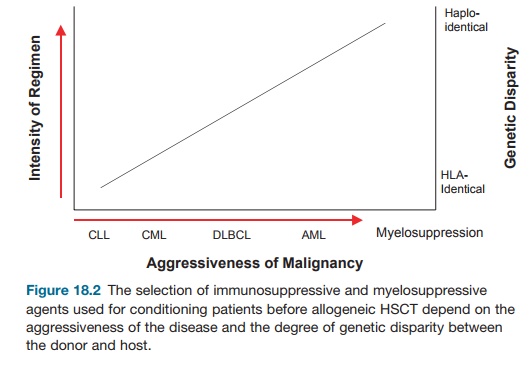
Graft-versus-Host Disease
The MHC constitutes one of the most highly polymorphic sets of genes known. From the standpoint of normal biology, this provides a species with a survival advan-tage by conferring the ability to respond to a myriad of potentially harmful peptide antigens that can be presented by a variety of MHC molecules. This polymorphism is, however, one of the major impediments to allografting because of the risk of GvHD, which is a unique complication of alloge-neic HSCT. The organ to be transplanted has immunologic capacity. Hence, one can turn the process of solid organ graft rejec-tion inside out to conceptualize what occurs during a GvH reaction. During GvHD, the engrafted, immunocompetent donor cells recognize the host as foreign and mount an immunologic attack.
The dominant immunologic factors underlying GvHD are the HLA compat-ibility between donor and recipient and the presence of immunocompetent T cells in the graft. GvHD occurs even between fully HLA-identical siblings, however, because nucleotide polymorphisms dif-fer between such individuals, which are processed and presented by MHC as self-antigens (Figure 18.3). These so-called minor histocompatibility antigens actually require MHC identity between siblings, as this ensures recognition of MHC bearing self-antigen by the correct T-cell recep-tor in the donor T-cell repertoire. In cases of MHC disparity, which are sometimes used in the absence of a fully matched donor, GvH would additionally target the full alloantigenic disparities themselves. Target organs include those in which anti-gen-presenting cells reside like the skin, gut, liver, and lung.
There are two forms of GvHD, acute and chronic. Acute GvHD is primarily T-cell and cytokine mediated, whereas chronic GvHD is thought to be B-cell and antibody mediated. Classic acute GvHD typically occurs within the first 100 days after allogeneic HSCT; but persistent, recurrent, or late-onset forms of acute
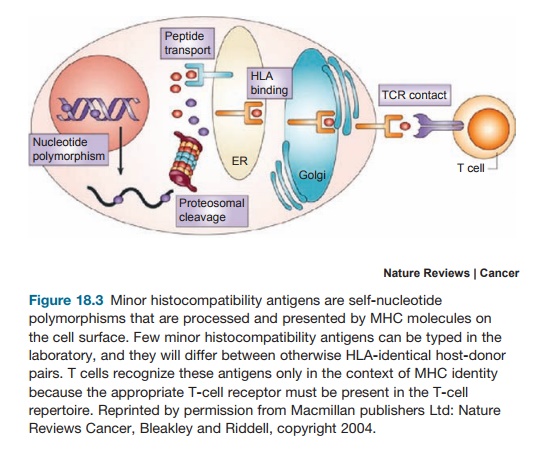
GvHD are more frequently recognized beyond this time point with the increas-ing use of nonmyeloablative regimens and unrelated donor sources. Chronic GvHD usually occurs beyond 100 days, but an overlap syndrome with features of acute GvHD is also now recognized. Clinical manifestations of acute GvHD can run the gamut from mild skin rash, anorexia, mild diarrhea, or low-grade cholestasis to full-thickness skin sloughing, liters of bloody diarrhea, and severe liver dysfunction with lethal consequences. Chronic GvHD resembles autoimmune diseases, for exam-ple, scleroderma, in many of its manifes-tations. To the extent that DNA-based HLA typing is now more precise and the therapeutic measures available for pre-vention and treatment of GvHD are more varied, though still imperfect, death due solely to GvHD should be an exceptionally rare event. Treatment of GvHD, however, can involve increasing layers of immune suppression, which can result in serious morbidity or even death.
Unmodified PBSCs seem to carry a greater risk of GvHD, especially chronic, than do bone marrow allografts. The basis for this is largely speculative but remains an area of active investigation. PBSGs are composed of a variety of cellular sub-sets that may have their own influence on transplant biology. Large numbers of T cells in the PBSC allografts may also increase GvHD risk. This seems unlikely as the sole explanation, however, because when controlled for T-cell numbers, the acute GvHD risk may actually be lower after unmodified PBSC than after unmod-ified bone marrow allografting. Interest-ingly, mobilized PBSC grafts induce less acute GvHD than grafts composed of steady state peripheral blood cells, sug-gesting that there are qualitative differ-ences in T-cell subsets that may carry greater importance than T-cell numbers. UCB does not contain mature or mem-ory T cells. The risk of GvHD mediated by naïve donor T cells sensitized against the host, while not zero, is therefore still considerably less than that which occurs after transplantation of other unmodified transplant products, even for a greater degree of host-donor mismatch.
Prevention of GvHD requires either depletion of T cells from the allograft or pharmacologic prophylaxis to limit sen-sitization of donor T cells against host antigens. Drugs for GvHD prophylaxis include those used for prevention of solid organ graft rejection such as cyclospo-rine, tacrolimus, sirolimus, mycopheno-lic acid, and methotrexate (Figure 18.4). Corticosteroids are used acutely for treat-ment of GvHD if it occurs; but there are no proven therapies for steroid-refrac-tory GvHD, whether acute or chronic. T-cell depletion is extremely effective in reducing acute GvHD and is more effec-tive in preventing chronic GvHD than is pharmacologic prophylaxis. Techniques to decrease the number of T cells in the HSC allograft have been developed for application in vitro and in vivo. Methods that achieve less T-cell depletion than others may sometimes require pharmaco-logic GvHD prophylaxis as well.
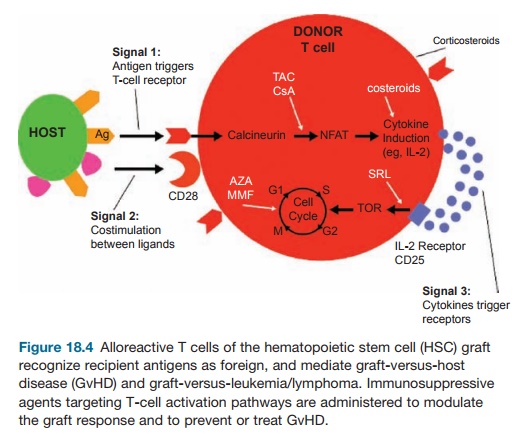
Graft-versus-Host Disease and Graft-versus-Leukemia/Lymphoma
It was well after transplants were under-taken in humans that seminal work by Korngold and Sprent in the late 1970s and early 1980s established that donor T cells mediated the onset of acute GvHD between mice that were MHC identical but mismatched for minor histocompat-ibility antigens. We also now know that T cells are important for the GvL effect. In fact, donor leucocyte infusions (DLI) after allogeneic HSCT can often eradicate minimal residual disease recurrences and convert mixed-to full-donor chimeras. These DLIs are dosed according to their T-cell contents; but the products are usually not purified T cells and therefore contain additional potential effector populations
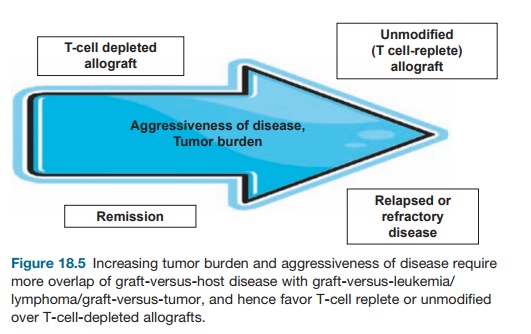
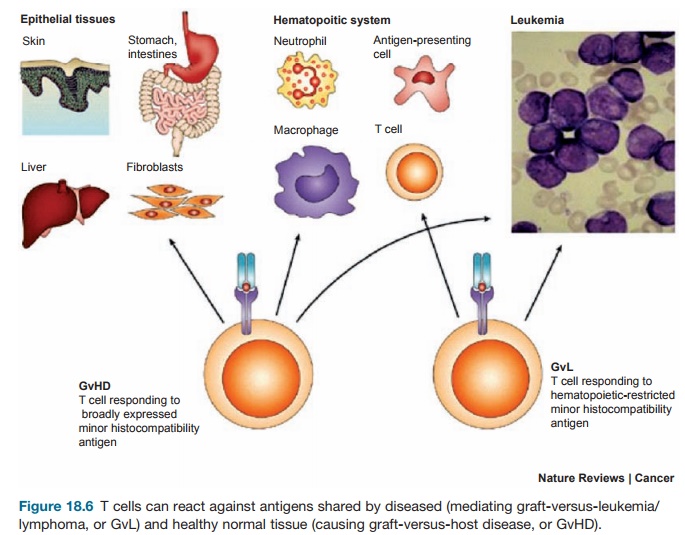
GvHD and GvL are discrete syndromes that may be conceptualized as two circles in a Venn diagram with variable degrees of overlap. Patients who are in complete remission (CR) at the time of transplant, can often receive T-cell-depleted allografts without developing GvHD and suffer no increased risk of relapse (Figure 18.5). In this case, GvL can be easily separated from GvHD. The higher the disease burden going into transplant, however, the higher the acceptable risk for GvHD to maintain the desired GvL effect. Patients who are not in CR at the time of transplant there-fore require more overlap between these two processes. Such patients may there-fore require a T-cell replete or unmodified allograft, accepting the risk of inducing GvHD to ensure sufficient GvL. The holy grail, however, is to identify antigens that are unique to the malignancy and thus not shared by other target organs (Fig-ure 18.6). This is a formidable challenge because many if not most tumor antigens are self-antigens or differentiation antigens expressed by normal tissue. In the case of hematopoietic malignancies, destruc-tion of normal hematopoiesis when tar-geting unique leukemia-specific antigens is less problematic when hematopoiesis will be replaced anyway by a functioning allograft.
Related Topics