Chapter: Biochemistry: Inborn Errors of Metabolism
Galactosemia: Causes, Symptoms
Galactosemia
It is an inherited disorder, in which there is
inability to convert galactose to glucose in a normal manner. The incidence of
this disease is about 1 in 18,000 live births.
1. Causes
Enzyme deficiency in galactose metabolic pathway
causes galactosemia. The pathway for conversion of galactose to glucose is
shown in figure 7.1.
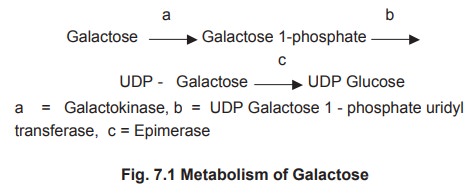
In galactosemia, there is inability to
metabolize galactose which may be caused by the enzyme deficiency of a =
galactokinase, b = UDP Galactose 1 - phosphate uridyl transferase.
2. Symptoms
The deficiency of galactose 1 - phosphate uridyl
transferase is clinically important. Due to the enzyme defect galactose
accumulates in blood and is reduced by aldose reductase in the eye to the
corresponding galactitol which causes cataract.
The general condition is more severe if it is
due to a defect in galactose 1 - phosphate uridyl tranferase, since galactose
1- phosphate accumulates and depletes the liver of inorganic phosphate.
Ultimately, liver failure and mental deterioration results.
Infants appear normal at birth but later they
show failure to thrive and become lethargic. They have frequent vomitting and
hypoglycemia. After 2 - 3 months of age the liver may show fatty infiltration
and lead to cirrhosis (non functioning of liver cells). Galactosemia at this
age is associated with mental retardation due to accumulation of galactose and
galactose 1 - phosphate in cerebral cortex. So, the galactosemic child fails to
grow and suffer from liver damage and mental retardation.
Related Topics