Chapter: Modern Pharmacology with Clinical Applications: Drugs Used in Gastrointestinal Disorders
Drugs that Decrease or Neutralize Gastric Acid Secretion
DRUGS THAT
DECREASE OR NEUTRALIZE GASTRIC ACID SECRETION
Functionally, the gastric
mucosa is divided into three ar-eas of secretion. The cardiac gland area secretes mucus and pepsinogen. The oxyntic (parietal) gland area, which
corresponds to the fundus and body of the stomach, se-cretes hydrogen ions,
pepsinogen, and bicarbonate. The pyloric
gland area in the antrum secretes gastrin and mucus.
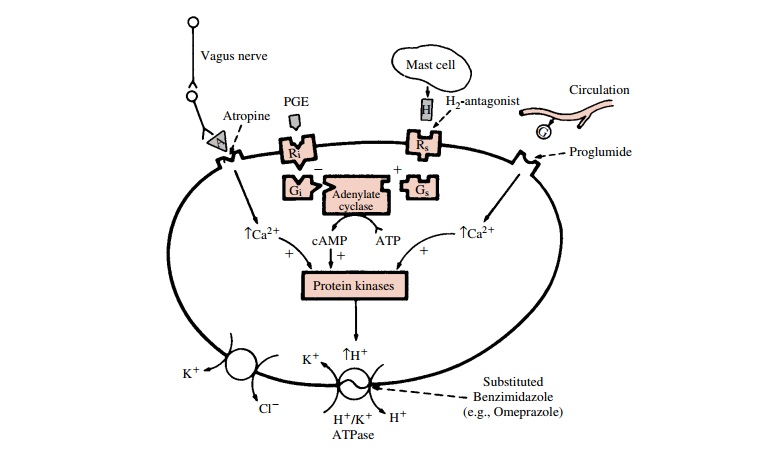
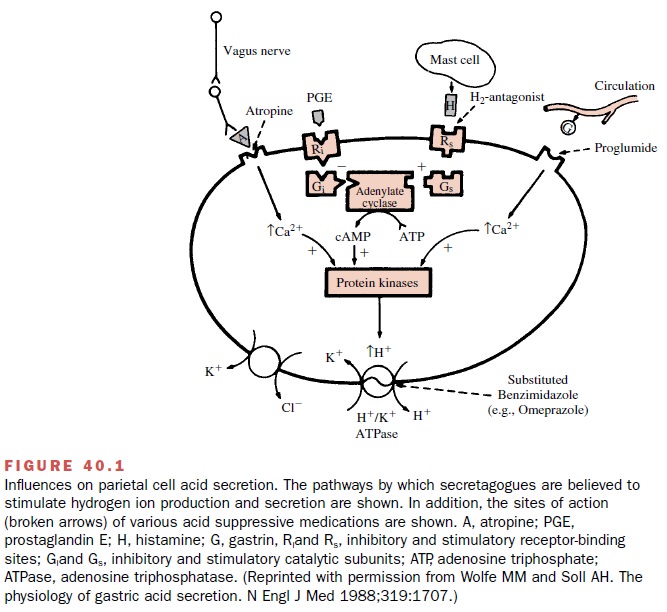
The parietal cells secrete H+
in response to gastrin, cholinergic, and histamine stimulation (Fig. 40.1).
Both cholinergic- and gastrin-induced types of stimulation bring about a
receptor-mediated rise in intracellular cal-cium, an activation of
intracellular protein kinases, and eventually an increased activity of the H+
–K+ pump leading to acid secretion into the gastric lumen.
Follow-ing histamine stimulation, a guanine nucleotide–binding protein (Gs)
activates adenylyl cyclase, leading to an in-crease in intracellular levels of
the second messenger, cyclic adenosine monophosphate (cAMP). Activation of
cAMP-dependent protein kinases initiates the stimu-lation of the H+ –K+
pump.
The cephalic–vagal axis, gastric distention, and local mucosal chemical receptors can modulate acid secre-tion by the stomach. The smell, taste, sight, or discussion of food may result in cephalic–vagal postganglionic cholinergic stimulation of target parietal cells and en-hanced antral gastrin release. After food is ingested, gastric distention initiates vagal stimulation and short intragastric neural reflexes, both of which increase acid secretion. Proteins in ingested meals also stimulate acid secretion. Evidence from animal studies suggests that after protein amino acids are converted to amines, gas-trin is released.
Gastric acid secretion is
inhibited in the presence of acid itself. A negative feedback occurs when the
pH ap-proaches 2.5 such that further secretion of gastrin is in-hibited until
the pH rises. Ingested carbohydrates and fat also inhibit acid secretion after
they reach the intes-tines; several hormonal mediators for this effect have
been proposed. The secretion of pepsinogen appears to parallel the secretion of
H+ , while the patterns of secre-tion of mucus and bicarbonate have
not been well char-acterized.
The integrity of the mucosal
lining of the stomach and proximal small bowel is in large part determined by
the mucosal cytoprotection provided by mucus and bi-carbonate secretion from
the gastric and small bowel mucosa. Mucus retards diffusion of the H+
from the gas-tric lumen back into the gastric mucosal surface. In ad-dition,
the bicarbonate that is secreted into the layer be-tween the mucus and
epithelium permits a relatively high pH to be maintained in the region next to
the mu-cosal surface. If any H+ does diffuse back to the level of
the mucosal surface, both the
local blood supply and the ability of the local cells to buffer this ion will
ultimately determine whether peptic ulceration will occur. With duodenal and
gastric peptic ulcer disease, a major causative cofactor is the presence of
gastric Helico-bacter pylori infection.
Medications that raise
intragastric pH are used to treat peptic ulcer disease and gastroesophageal
reflux disease. In addition, agents that enhance mucosal cyto-protection are
used to decrease ulcer risk.
Antacids
The rationale for the use of
antacids in peptic ulcer dis-ease lies in the assumption that buffering of H+
in the stomach permits healing. The use of both low and high doses of antacids
is effective in healing peptic ulcers as compared with placebo. Healing rates
are comparable with those observed after the use of histamine (H2)
blocking agents. The buffering agents in the various antacid preparations
consist of combinations of ingredi-ents that include sodium bicarbonate,
calcium carbon-ate, magnesium hydroxide, and aluminum hydroxide. If diarrhea
occurs or if there is renal failure, a magnesium-based preparation should be
discontinued. The agents are generally safe, but some patients resist because
some of the formulations are unpalatable and expensive.
A variety of adverse effects
have been reported fol-lowing the use of antacids. If sodium bicarbonate is
ab-sorbed, it can cause systemic alkalization and sodium overload. Calcium
carbonate may induce hypercalcemia and a rebound increase in gastric secretion
secondary to the elevation in circulating calcium levels. Magnesium hydroxide
may produce osmotic diarrhea, and the ex-cessive absorption of Mg in patients
with renal failure may result in central nervous system toxicity. Aluminum
hydroxide is associated with constipation; serum phos-phate levels also may
become depressed because of phosphate binding within the gut. The use of
antacids in general may interfere with the absorption of a number of
antibiotics and other medications.
H2-Receptor Antagonists
The histamine receptor
antagonists (H2 blockers) mar-keted in the United States are
cimetidine (Tagamet), ranitidine (Zantac), famotidine (Pepcid), and nizatidine (Axid). These agents bind to the H2-receptors
on the cell membranes of parietal cells and prevent histamine-induced
stimulation of gastric acid secretion. After pro-longed use, down-regulation of
receptor production oc-curs, resulting in tolerance to these agents. H2-blockers
are approved for the treatment of gastroesophageal re-flux disease, acute ulcer
healing, and post–ulcer healing maintenance therapy. Although there are
substantial differences in their relative potency, 70 to 85% of duo-denal
ulcers are healed during 4 to 6 weeks of therapy with any of these agents. The
incidence of healing of gastric ulceration after 6 to 8 weeks of therapy
ap-proaches 60 to 80% with the use of cimetidine or raniti-dine. Since
nocturnal suppression of acid secretion is particularly important in healing,
nighttime-only dosing can be used. Most are available in low-dose
over-the-counter formulations.
Cimetidine, the first
released H2-blocker, like hista-mine, contains an imidazole ring
structure. It is well ab-sorbed following oral administration, with peak blood
levels 45 to 90 minutes after drug ingestion. Blood lev-els remain within
therapeutic concentrations for ap-proximately 4 hours after a 300-mg dose.
Following oral administration, 50 to 75% of the parent compound is excreted
unchanged in the urine; the rest appears pri-marily as the sulfoxide
metabolite.
Cimetidine may infrequently
cause diarrhea, nau-sea, vomiting, or mental confusion. A rare association with
granulocytopenia, thrombocytopenia, and pancy-topenia has been reported.
Gynecomastia has been demonstrated in patients receiving either high-dose or long-term
therapy. This occurs because cimetidine has a weak antiestrogen effect. Since
cimetidine is partly me-tabolized by the cytochrome P450 system,
coadminis-tered drugs such as the benzodiazepines, theophylline, and warfarin,
which are also metabolized by this system, may accumulate if their dosage is
not adjusted.
Ranitidine is well absorbed
after oral administra-tion, with a peak plasma level achieved 1 to 3 hours
af-ter ingestion. Elimination is by renal (25%) and hepatic (50%) routes. The
half-life of elimination is 2.5 to 3.0 hours. Nizatidine is the newest H2-receptor
antagonist. Similar to ranitidine, it has a relative potency twice that of
cimetidine. About 90% of an oral dose is absorbed, with a peak plasma
concentration occurring after 0.5 to 3 hours; inhibition of gastric secretion
is present for up to 10 hours. The elimination half-life is 1 to 2 hours, and
more than 90% of an oral dose is excreted in the urine. Famotidine has an onset
of effect within 1 hour after oral administration, and inhibition of gastric
secretion is present for the next 10 to 12 hours. It is the most potent H2-blocker.
Elimination is by renal (65–70%) and he-patic (30–35%) routes. Ranitidine,
famotidine, and niza-tidine do not alter the microsomal cytochrome P450
metabolism of other drugs, nor do they cause gyneco-mastia. A reduction in
dosage of any of the H2-blockers is recommended in the presence of
renal insufficiency.
Proton Pump Inhibitors
The proton pump inhibitors
available in the United States are omeprazole (Prilosec), lansoprazole (Prevacid),
pan-toprazole (Protonix), rabeprazole
(Aciphex), and es-omeprazole (Nexium). These are substituted
benzimida-zole prodrugs, which accumulate on the luminal side of parietal
cells’ secretory canaliculi. They become acti-vated by acid transport and then
bind covalently to the actual H+ –K ATPase enzymes (proton pumps)
irreversibly blocking them. These drugs
markedly inhibit gastric acid
secretion. New proton pumps are continu-ously formed, and thus no tolerance
develops. Peptic ulcers and erosive esophagitis that are resistant to other
therapies will frequently heal when these agents are used. The proton pump
inhibitors are also used to treat patients with Zollinger-Ellison syndrome, which is the result of a
gastrin-hypersecreting neuroendocrine tumor.
The prodrugs are unstable in
the presence of acid and therefore must be administered as an enteric-coated
preparation or as a buffered suspension. Pantoprazole is also available in an
intravenous formulation. The most commonly reported side effects are diarrhea
and headache. Hypergastrinemia has been noted as a reac-tion to the marked
reduction in acid secretion. Gastric carcinoid tumors have developed in rats
but not in mice or in human volunteers, even after long-term use.
Related Topics