Chapter: Medical Microbiology: An Introduction to Infectious Diseases: Introduction to Pathogenic Parasites: Pathogenesis and Chemotherapy of Parasitic Diseases
Drugs - Parasites Chemotherapy
Drugs
Heavy Metals
Arsenic and antimonial compounds have been used since ancient times. They form stable complexes with sulfur compounds and probably exert their biological effects by binding to sulfhydryl groups. They are toxic to the host as well as to the parasite and have their greatest impact on cells that are most metabolically active such as neuronal, renal tubular, intestinal epithelial, and bone marrow stem cells. Their differential toxicity and therapeu-tic value are due to enhanced uptake by the parasite and its intense metabolic activity. Only one trivalent arsenical, melarsoprol* (Mel B), is now widely used. It is capable of penetrating the blood–brain barrier and is effective in all stages of trypanosomiasis. Because of its toxicity, it is employed only when less toxic agents have failed or the central nervous system is involved. The recently introduced less toxic trypanocides that penetrate the blood–brain barrier may soon replace this drug.
Antimonial agents are now restricted to the management of leishmanial infections. Two pentavalent compounds, sodium stibogluconate* (Pentostam) and meglumine antimoniate†(Glucantime), are used for all forms of leishmaniasis. In disseminated disease, prolonged therapy is usually required and relapses often occur. In localized cutaneous leishmaniasis, cure is usually achieved with a relatively brief course. Toxic side effects are similar to those of the arsenicals.
Antimalarial Quinolines
Cinchona bark was used in Europe for the treatment of fever as early as 1640. Only after Pelletier and Caventou isolated quinine from cinchona in 1820 did this alkaloid gain widespread acceptance as an antimalarial. Synthesis of new quinolines was stimulated by the interruption of quinine supplies during World Wars I and II and, after 1961, by the growing impact of drug-resistant falciparum malaria in several areas of the world. Among the most effective agents are those that share the double-ring structure of quinine.
Current analogs fall into three major groups: 4-aminoquinolines, 8-aminoquinolines, and 4-quinolinemethanols. Selective destruction of intracellular parasites results from ac-cumulation of the quinolines by parasitized host cells. Most of these agents appear to block nucleic acid synthesis by intercalation into double-stranded DNA. However, the failure of the 4-quinolinemethanols to intercalate indicates that other mechanisms, per-haps inhibition of heme polymerase, with the build up of toxic hemoglobin metabolites within the malarial parasite, are involved.
Quinine, 4-aminoquinolines, and 4-quinolinemethanols are preferentially concen-trated in parasitized erythrocytes and rapidly destroy the erythrocytic stage of the parasite that is responsible for the clinical manifestations of malaria. Thus, these agents can be used either prophylactically to suppress clinical illness should infection occur or thera-peutically to terminate an acute attack. They do not concentrate in tissue cells, and thus organisms sequestered in exoerythrocytic sites, particularly the liver, survive and may later reestablish erythrocytic infection and produce a clinical relapse. The 8-aminoquino-lines accumulate in tissue cells, destroy hepatic parasites, and effect a radical cure.
Chloroquine phosphate, a 4-aminoquinoline, is the most widely used of the blood schizonticidal drugs. In the doses used for long-term malarial prophylaxis, it has proven remarkably free of untoward effects. Primaquine phosphate, the 8-aminoquinoline used to eradicate persistent hepatic parasites, has toxic effects related to its oxidant activity. Methemoglobinemia and hemolytic anemia are particularly frequent in patients with glu-cose-6-phosphate dehydrogenase deficiency, because they are unable to generate suffi-cient quantities of the reduced form of nicotinamide adenine dinucleotide to respond to this oxidant stress. Typically, the anemia is severe in patients of Mediterranean and Far Eastern ancestry and mild in black patients.
Quinine is the most toxic of the quinolines and is currently used primarily to treat the strains of P. falciparum resistant to several blood schizonticidal agents that are spreading rapidly through Asia, Latin America, and Africa. Chloroquine resistance is the most fre-quent and worrisome, because suitable alternatives to this safe and highly effective agent are few. The mechanism of resistance is not clearly understood, but resistant organisms fail to accumulate chloroquine. Experimental reversal of resistance with calcium channel blockers suggests that the failure to accumulate this agent results from a rapid release mechanism. Quinidine, a less cardiotoxic optical isomer of quinine, is more readily available in the United States and is preferred to quinine when parenteral administration is required. Meflo-quine, a more recently developed oral 4-quinolinemethanol, originally displayed a high level of activity against most chloroquine-resistant parasites; however, mefloquine-resistant strains of P. falciparum are now widespread in Southeast Asia, and present, to a lesser de-gree, in South America. Resistant strains have recently been identified in Africa.
Phenanthrene methanols are not, in the strict sense, quinine analogs. Nevertheless, they are structurally similar to this group of agents and, together with them, were discov-ered to have antimalarial activity during the second World War. Halofantrine†, the most effective of the group, has only recently become available. In vitro and in vivo studies demonstrated that it is an effective blood schizonticide against both sensitive and mul-tidrug-resistant strains of P. falciparum. Its mechanism of action was originally thought to differ from that of quinine and mefloquine. Recently, mefloquine-resistant strains of P. falciparum have demonstrated decreased sensitivity to halofantrine, raising the possi-bility of cross-resistance between these two agents. Rarely, halofantrine has produced fa-tal heart arrhythmias, and it should not be given to patients with cardiac conduction abnormalities. It is otherwise well tolerated and appears to be free of teratogenicity. Oral absorption is both slow and erratic, reaching maximum concentrations in 5 to 7 hours; its half-life is relatively short (1 to 3 days). Clinical studies have demonstrated high failure rates when the drug is given in a single dose; cure rates with multiple-dose regimens, however, have been high.
Quinones
Atovaquone is a novel hydroxynaphthoquinone that shows promise in the treatment of malaria and toxoplasmosis. In the search for effective antimalarial agents during World War II, a number of hydroxynaphthoquinones were found to have antimalarial activity in experimental animals; however, all were rapidly metabolized in humans and proved inef-fective in the treatment of malarious patients. In the 1980s a single hydroxynaphtho-quinone, atovaquone, was found to be both highly effective in vitro against P. falciparum and metabolically stable in humans when administered orally. Its antiparasitic activity appears to result from the specific blockade of pyrimidine biosynthesis secondary to the inhibition of the parasite’s mitochondrial electron transport chain at the ubiquinol– cytochrome c reductase region (complex III). Its long half-life (70 hours) and lack of seri-ous adverse reactions suggested that it would be of great value in the treatment of malaria. Efficacy trials established its capacity to effect rapid clearance of parasitemia in patients with chloroquine-resistant falciparum malaria. Frequent parasitic recrudescences were eliminated when atovaquone was administered in combination with proguanil or tetracycline. Subsequently, this agent has shown to be effective for the treatment of toxo-plasmosis in patients with acquired immunodeficiency syndrome (AIDS). Unlike other antitoxoplasma agents, atovaquone has been found to be active against T. gondii cysts as well as tachyzoites, suggesting this agent may produce radical cure. Supporting this is the infrequency with which cessation of atovaquone treatment of toxoplasmic cerebritis in AIDS patients has resulted in relapse. Relapse following atovaquone treatment of pneu-mocystosis in this same patient population appears similarly uncommon.
Folate Antagonists
Folic acid serves as a critical coenzyme for the synthesis of purines and ultimately DNA. In protozoa, as in bacteria, the active form of folic acid is produced in vivo by a simple two-step process. The first, the conversion of para-aminobenzoic acid to dihydrofolic acid, is blocked by sulfonamides. The second, the transformation of dihydro- to tetrahy-drofolic acid, is inhibited by folic acid analogs (folate antagonists), which competitively inhibit dihydrofolate reductase. Used together with sulfonamides, folate antagonists are very effective inhibitors of protozoan growth.
Trimethoprim, an inhibitor of dihydrofolate reductase, is used in combination with sulfamethoxazole to treat toxoplasmosis. Another folate antagonist, pyrimethamine, has a high affinity for sporozoan dihydrofolate reductase and has been particularly effective, when used with a sulfonamide, in the management of clinical malaria and toxoplasmosis.
In East Africa, a third folate antagonist, proguanil, is commonly taken in combination with chloroquine for malaria prophylaxis. Acquired protozoal resistance to folate antagonists is mutational and generally has been limited to particular species of malarial parasites.
Folate antagonists may result in folate deficiency in individuals with limited folatereserves, such as newborns, pregnant women, and the malnourished. This is of great concern when large doses are used for prolonged periods, as in the treatment of acute toxoplasmosis. When folate antagonists are used with sulfonamides, the entire range of sulfonamide toxic effects may be seen. Patients with AIDS appear to suffer an unusually high incidence of toxic side effects to trimethoprim–sulfamethoxazole.
Qinghaosu (Artemisinin†)
This natural extract of the plant Artemisia annua (qing hao, sweet wormwood) is a sesquiterpenelactone peroxide that is structurally distinct from all other known antiparasitic compounds. Extracts of qing hao were recommended for the treatment of fevers in China as early as AD 341; their specific antimalarial activity was defined in 1971. Although qinghaosu has also been shown to be active against the free-living ameba Naegleria fowleria and several trematodes, including Schistosoma japonicum, Schistosoma mansoni, and Clonorchis sinensis, its greatest impact to date has been in the treatment of malaria. Extensive investigations showed it to be schizonticidal for both chloroquine-sensitive and chloroquine-resistant strains of P. falciparum. Several derivatives, among them artemether†and artesunate, are significantly more active than the parent compound. All are concentrated in parasitized erythrocytes where they decompose, releasing free radicals, which are thought to be damaging to parasitic membranes. Artemisinin compounds act more rapidly than other antimalarial agents, stopping parasite development and preventing cytoadherence in falciparum malaria. Although depression of reticulocyte counts has been noted, these agents appear significantly less toxic than quinoline antimalarials. As there is some evidence that they may possess teratogenic properties, they should not be used in pregnancy. Importantly, they may be given orally, rectally (by suppository), and parenterally. Relapses can occur unless they are given for several days or combined with a second agent such as mefloquine or tetracycline.
Nitroimidazoles
Metronidazole, a nitroimidazole, was introduced in 1959 for the treatment of trichomonia-sis. Subsequently, it was found to be effective in the management of giardiasis, amebiasis, and a variety of infections produced by obligate anaerobic bacteria. Energy metabolism in all of them depends on the presence of low-redox-potential compounds, such as ferre-doxin, to serve as electron carriers. These compounds reduce the 5-nitro group of the imi-dazoles to produce intermediate products responsible for the death of the protozoal and bacterial cells, possibly by alkylation of DNA. Resistance, although uncommon, has been noted in strains of T. vaginalis lacking nitroreductase activity. Of greater concern is in vitro evidence of mutagenicity. Metronidazole is the drug of choice for trichomoniasis and invasive amebiasis. It is effective in giardiasis although not yet approved by the Food and Drug Administration for use in this infection. Tinidazole, a newer nitroimidazole not yet available in the United States, appears to be both a more effective and less mutagenic antiprotozoal agent. Its greater lipid solubility improves cerebrospinal fluid levels and in vitro activity.
Benzimidazoles
As the name benzimidazole implies, the basic structure of these antiparasitic agents con- sists of linked imidazole and benzene rings. Unlike their antiprotozoal cousins discussedabove, the benzimidazoles are broad-spectrum anthelmintic agents. The prototype drug, thiabendazole, acts against both adult and larval nematodes and was shown to be useful in the management of cutaneous larva migrans, trichinosis, and most intestinal nematode in-fections soon after its introduction in the early 1960s. The mechanism by which it exerts its anthelmintic action is uncertain. It is known to inhibit fumarate reductase, an impor-tant mitochondrial enzyme of helminths. The primary mode of action, however, may derive from the known capacity of all benzimidazoles to inhibit the polymerization of tubulin, the eukaryotic cytoskeletal protein, as described for mebendazole below. Side ef-fects are mild, related to the gastrointestinal tract or liver, and rapidly disappear with the discontinuation of the drug. Hypersensitivity reactions, induced either by the drug or by antigens released from the damaged parasite, may occur.
Mebendazole, a carbamate benzimidazole introduced in 1972, has a spectrum similar to that of thiabendazole, but also has been found to be effective against a number of ces-todes, including Taenia, Hymenolepsis, and Echinococcus. It irreversibly blocks glucose uptake of both adult and larval worms, resulting in glycogen depletion, cessation of ATP formation, and paralysis or death. It does not appear to affect glucose metabolism in hu-mans and is thought to exert its effect in worms by binding to tubulin, thus interfering with the assembly of cytoplasmic microtubules, structures essential to glucose uptake. Unlike thiabendazole, the drug is not well absorbed from the gastrointestinal tract and may owe part of its effectiveness against intestine-dwelling adult worms to its high con-centrations in the gut. Toxicity is uncommon. Teratogenic effects have been observed in experimental animals; its use in infants and pregnant women is contraindicated.
Albendazole is a benzimidazole carbamate that has recently been made available in the United States. It has a somewhat broader spectrum than that of its close relative, mebendazole, being more active against Strongyloides stercoralis and several tissue nematodes. In addition to the vermicidal and larvicidal properties that it shares with other benzimidazoles, it is ovicidal, enhancing its effectiveness in tissue cestode infec-tions such as echinococciasis and cysticercosis. Its activity against Giardia, one of the most common intestinal protozoa, makes it an appealing candidate for the treatment of polyparasitism. Although it shares the teratogenic potential of other benzimidazoles, it is otherwise extremely well tolerated. Single-dose therapy is effective in the management of many intestinal nematode infections.
Avermectins
Avermectins are macrocyclic lactones produced as fermentation products of Streptomycesavermitilis. Structurally similar to the macrolide antibiotics, they are effective at extremelylow concentration against a wide variety of nematodes and arthropods. The avermectins appear to induce neuromuscular paralysis by acting on a receptor of the parasites-peripheral neurotransmitter, gamma-aminobutyric acid (GABA). In mammals, GABA is confined to the central nervous system, and because the avermectins do not cross the blood–brain barrier in significant concentration, they do not appear to produce significant untoward effects in the mammalian host. Ivermectin, a derivative of avermectin B1, is currently the drug of choice for the treatment of onchocerciasis and is undergoing evalua-tion for the treatment of other human filarial infections. Its usefulness in other parasitic infections of humans remains to be established.
Praziquantel
Praziquantel, a heterocyclic pyrazinoisoquinoline, is an important new anthelmintic effec-tive against a broad range of cestodes and trematodes, many of which had been poorly re-sponsive to previously available agents. It is given in one to three doses. The drug is rapidly taken up by susceptible helminths, in which it appears to induce the loss of intra-cellular calcium, tetanic muscular contraction, and destruction of the tegument. The dif-ferential toxicity of this agent may be related to the inability of susceptible worms to metabolize the drug. Aside from transient, mild gastrointestinal symptoms, praziquantel appears remarkably free of side effects in humans. It is currently the drug of choice for the treatment of schistosomiasis, clonorchiasis, opisthorchiasis, and neurocysticercosis.
Good activity has been demonstrated against other common trematode and cestode infec-tions. Its high level of safety suggests that it may well play a significant role in worldwide mass therapy campaigns.
Eflornithine† (Difluoromethylornithine)
Eflornithine is a specific, enzyme-activated, irreversible inhibitor of ornithine decarboxy-lase (ODC). In mammalian cells, decarboxylation of ornithine by ODC is a mandatory step in the synthesis of polyamines, compounds thought to play critical roles in cell divi-sion and differentiation. Originally developed as an antineoplastic agent, eflornithine proved ineffective in cancer chemotherapy trials. With the discovery that polyamines of Trypanosoma species were also synthesized from ornithine, eflornithine was successfullytested in the treatment of animal trypanosomiasis. Host survival was high and associated with decreases in parasitic polyamines and inhibition of nucleic acid synthesis. In the dosage required to treat trypanosomiasis, mammals tolerated the agent well, presumably because T. brucei is 100 times more sensitive to the effects of eflornithine than are mam-malian cells. Eflornithine appears to be cytostatic and requires an intact host immune sys-tem for maximum effect.
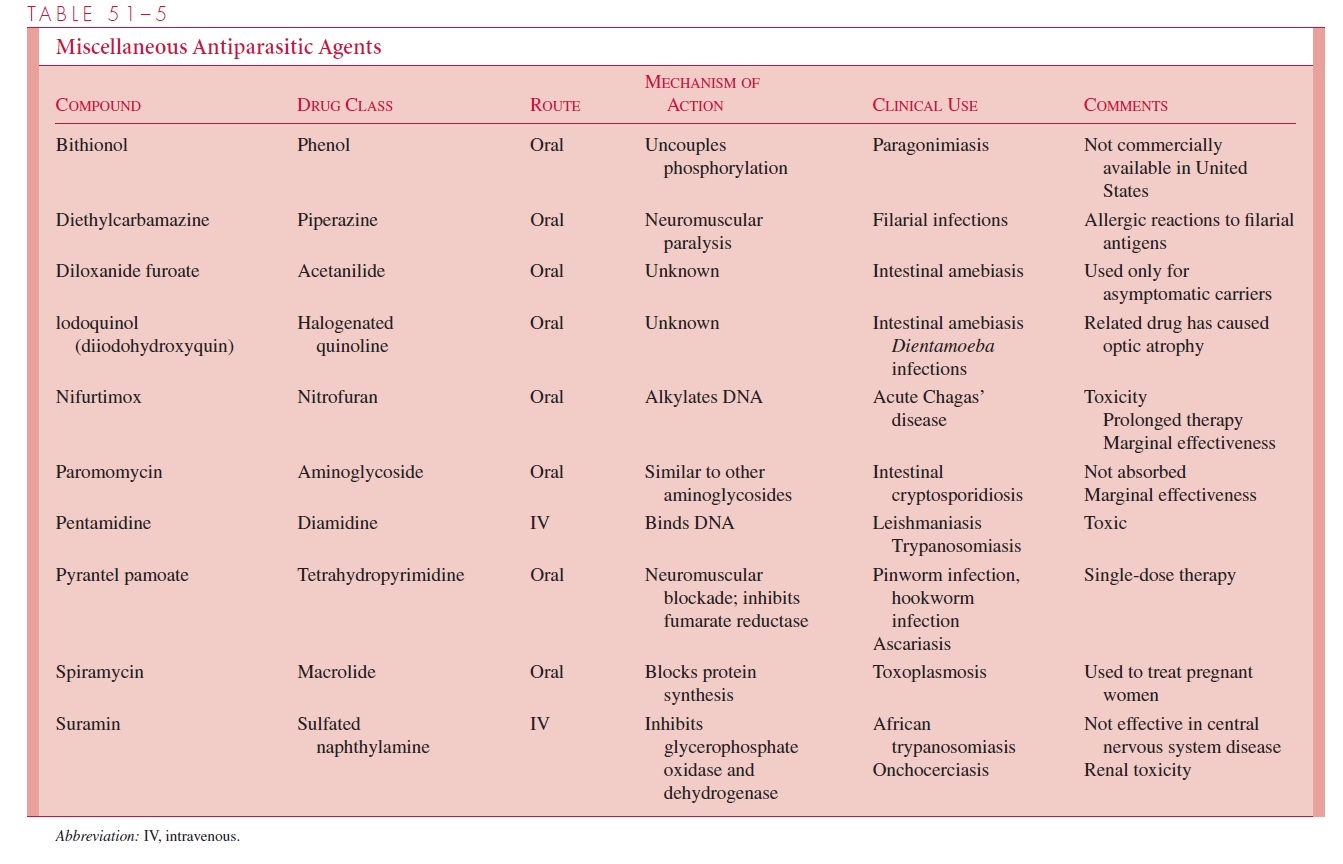
Other Antiparasitic Agents
A number of antiparasitic agents used in therapy, their properties, and their clinical uses are listed in Table 51–5.
Related Topics