Chapter: Modern Pharmacology with Clinical Applications: Directly and Indirectly Acting Cholinomimetics
Direct-Acting Parasympathomimetic Drugs
DIRECT-ACTING PARASYMPATHOMIMETIC
DRUGS
Acetylcholine
Acetylcholine is an ester of
choline and acetic acid, the prototype for a small family of choline ester
com-pounds. The choline moiety of ACh contains a quater-nary ammonium group
that gives ACh a permanent positive charge, making it very hydrophilic and
mem-brane impermeant.
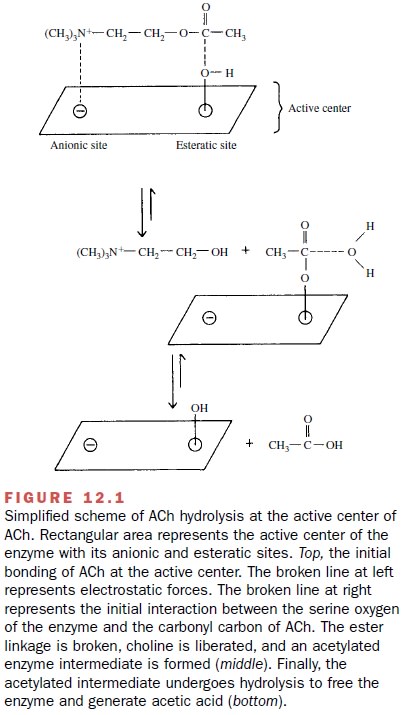
ACh is degraded by a group of
enzymes called cholinesterases. These enzymes catalyze the hydrolysis of ACh to
choline and acetic acid (Fig. 12.1). The active center of cholinesterase has
two areas that interact with ACh: the anionic site and the esteratic site. The
anionic site contains a negatively charged amino acid that binds the positively
charged quaternary ammonium group of ACh through coulombic forces. This
probably serves to bring the ester linkage of ACh close to the esteratic site
of the enzyme. The esteratic site contains a serine residue, which is made more
reactive by hydrogen bonding to a nearby histidine residue. The nucleophilic
oxygen of the serine reacts with the carbonyl carbon of ACh, thereby breaking
the ester linkage. During this re-action, choline is liberated and an
acetylated enzyme is formed. The latter intermediate is rapidly hydrolyzed to
release acetic acid and regenerate the active enzyme. The entire process takes
about 150 microseconds, one of the fastest enzymatic reactions known.
There are two major types of cholinesterases: acetylcholinesterase (AChE) and pseudocholinesterase (pseudo-ChE). AChE (also known as true, specific, or erythrocyte cholinesterase) is found at a number of sites in the body, the most important being the cholinergic neuroeffector junction. Here it is localized to the pre-junctional and postjunctional membranes, where it rap-idly terminates the action of synaptically released ACh. It is essential to recognize that the action of ACh is terminated only by its hydrolysis.
There is no reuptake sys-tem in cholinergic
nerve terminals to reduce the con-centration of ACh in a synaptic cleft, unlike
the reup-take systems for other neurotransmitters such as dopamine, serotonin,
and norepinephrine. Therefore, in-hibition of AChE can greatly prolong the
activation of cholinoreceptors by ACh released at a synapse.
Pseudo-ChE (also known as
butyryl-, plasma, and nonspecific cholinesterase) has a widespread
distribu-tion, with enzyme especially abundant in the liver, where it is
synthesized, and in the plasma. In spite of the abundance of pseudo-ChE, its
physiological function has not been definitively identified. It does, however,
play an important role in the metabolism of such clini-cally important
compounds as succinylcholine, pro-caine, and numerous other esters.
Derivatives of ACh: Methacholine, Carbachol, and Bethanechol
The therapeutic usefulness of
ACh is limited by (1) its lack of selectivity as an agonist for different types
of choli-noreceptors and (2) its rapid degradation by cholin-esterases. These
limitations have been circumvented in part by the development of three choline ester derivatives of ACh:
methacholine (Provocholine), carbachol (Isopto
Carbachol, Miostat) and bethanechol (Urecholine). Methacholine differs from ACh only in the addition of a methyl
group at the -carbon of ACh. This modification greatly increases its
selectivity for muscarinic receptors relative to nicotinic receptors, and it
renders methacholine resistant to the pseudo-ChE in the plasma and decreases
its susceptibility to AChE, thereby increasing its potency and duration of
action compared to those of ACh. Carbachol differs from ACh only in the
substitution of a carbamoyl group for the terminal methyl group of ACh. This
substitution makes carbachol completely resistant to degradation by
cholinesterases but does not improve its selectivity for muscarinic versus
nicotinic receptors. Bethanechol combines the addition of the methyl group and
the substitution of the terminal carbamoyl group, pro-ducing a drug that is a
selective agonist of muscarinic re-ceptors and is resistant to degradation by
cholinesterases.
All of these drugs are very
hydrophilic and mem-brane impermeant because they retain the quaternary
ammonium group of the choline moiety of ACh.
Pilocarpine is a naturally
occurring cholinomimetic alkaloid that is structurally distinct from the choline
es-ters. It is a tertiary amine that crosses membranes rela-tively easily.
Therefore, it is rapidly absorbed by the cornea of the eye, and it can cross
the blood-brain bar-rier. Pilocarpine is a pure muscarinic receptor agonist,
and it is unaffected by cholinesterases. Muscarine
is an alkaloid with no therapeutic use, but it can produce dangerous
cholinomimetic stimulation following inges-tion of some types of mushrooms
(e.g., Inocybes).
Basic Pharmacology of the Directly Acting Parasympathomimetic Drugs
Methacholine, bethanechol,
and pilocarpine are selec-tive agonists of muscarinic receptors, whereas
carbachol and ACh can activate both muscarinic and nicotinic re-ceptors.
However, at usual therapeutic doses, the effects of carbachol and ACh are entirely
due to the activation of muscarinic receptors. This apparent preference for
muscarinic receptors can be attributed to the greater ac-cessibility and
abundance of these cholinoreceptors compared with the nicotinic receptors.
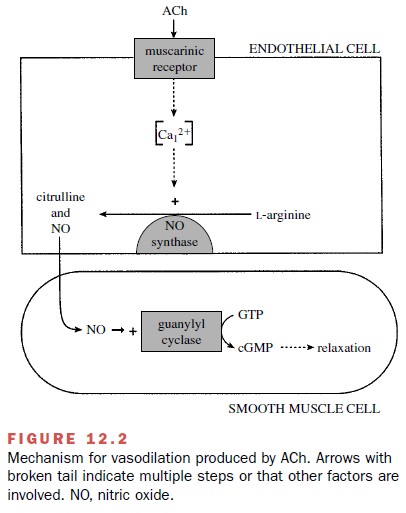
Cardiovascular Effects
Low doses of muscarinic
agonists given intravenously relax arterial smooth muscle and produce a fall in
bloodpressure. These responses result from the stimulation of muscarinic
receptors on vascular endothelial cells (Fig. 12.2). Activation of these
receptors causes the endothe-lial cells to synthesize and release nitric oxide.
Nitric ox-ide can diffuse into neighboring vascular smooth muscle cells, where
it activates soluble guanylyl cyclase, thereby increasing the synthesis of
cyclic guanosine monophos-phate (cGMP) and relaxing the muscle fibers. Most of
the resistance vasculature is not innervated by choliner-gic neurons, and the
physiological function of the en-dothelial muscarinic receptors is not known.
However, activation of these receptors by directly acting choli-nomimetic drugs
has major pharmacological signifi-cance, as the potentially dangerous
hypotension pro-duced by their activation is an important limitation to the
systemic administration of muscarinic agonists.
Although the release of ACh onto the heart by the vagus nerve slows the heart rate, a low dose of a mus-carinic agonist can sometimes increase the heart rate. This paradoxical effect is produced when the decrease in blood pressure produced by stimulation of endothe-lial muscarinic receptors, as described earlier, triggers the activation of a compensatory sympathetic reflex stimulation of the heart. Sympathetic stimulation in-creases heart rate and vasomotor tone, partially coun-teracting the direct vasodilator response. Therefore, the tachycardia produced by muscarinic agonists is indirect. At higher concentrations of a muscarinic agonist, the di-rect effects on cardiac muscarinic (M2) receptors in the SA node and A-V fibers become dominant.
Activation of M2 receptors increases the potassium permeability and
reduces cAMP levels, slowing the rate of depolar-ization and decreasing the
excitability of SA node and A-V fiber cells. This results in marked bradycardia
and a slowing of A-V conduction that can override the stim-ulation of the heart
by catecholamines released during sympathetic stimulation. In fact, very high
doses of a muscarinic agonist can produce lethal bradycardia and A-V block.
Choline esters have relatively minor direct effects on ventricular function,
but they can produce negative inotropy of the atria.
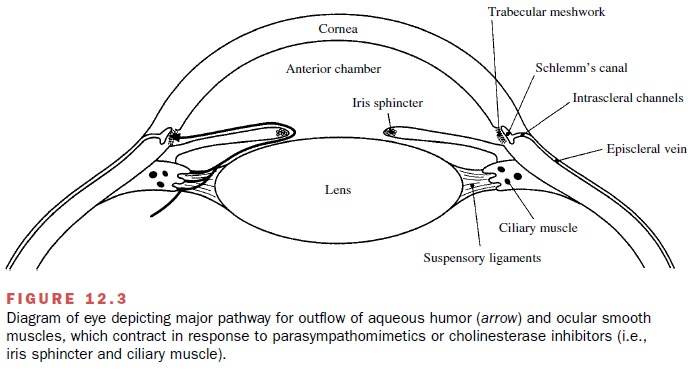
The Eye
When solutions of directly
acting cholinomimetics are applied to the eye (i.e., conjunctival sac), they
cause contraction of the smooth muscle in two important structures, the iris
sphincter and the ciliary muscles (Fig. 12.3). Contraction of the iris
sphincter decreases the di-ameter of the pupil (miosis). Contraction of the
circular fibers of the ciliary muscle, which encircles the lens, re-duces the
tension on the suspensory ligaments that nor-mally stretch and flatten the
lens, allowing the highly elastic lens to spontaneously round up and focus for
near vision (accommodation to near vision).
Other Organ Systems
Prominent effects within the
digestive tract include stimulation of salivation and acid secretion, increased
intestinal tone and peristaltic activity, and relaxation of most sphincters.
Bronchoconstriction and stimulation of secretions are prominent effects in the
respiratory sys-tem. Muscarinic agonists can also evoke secretion from
nasopharyngeal glands. Urination is promoted by stim-ulation of the detrusor
muscle of the bladder and is fa-cilitated by relaxation of the trigone and
external sphincter muscles.
Clinical Uses
Glaucoma
Cholinomimetic drugs are useful for treating glaucoma because they can decrease the resistance to the move-ment of fluid (aqueous humor) out of the eye (Fig. 12.3), thereby reducing the intraocular pressure. It is useful to distinguish between open-angle glaucoma, a chronic condition in which the porosity of the trabecular mesh-work is insufficient to permit the movement of fluid into the canal of Schlemm, and angle-closure glaucoma, an emergency condition in which an abnormal position of the peripheral iris blocks the access of fluid to the tra-becular meshwork. Open-angle glaucoma can be effec-tively treated with cholinomimetics such as pilocarpine and carbachol, because contraction of the ciliary muscle stretches the trabecular network, increasing its porosity and permeability to the outflow of fluid.
This beneficial effect, however, comes at the price of a
spasm of ac-commodation and miosis, which seriously disturb vi-sion.
Cholinomimetics, therefore, have been replaced by β-blockers and carbonic
anhydrase inhibitors, both of which decrease the formation of aqueous humor
with-out affecting vision. However, some patients simply do not respond to
these treatments or do not tolerate the cardiovascular side effects of the β-blockers,
and choli-nomimetics (most notably pilocarpine) remain as im-portant treatment alternatives.
Contraction of the iris
sphincter (miosis) by choli-nomimetic stimulation is less important than
contrac-tion of the ciliary muscle for treating angle-closure glau-coma, but it
may be essential as emergency therapy for acute-angle glaucoma to reduce
intraocular pressure prior to surgery (iridectomy). Contraction of the iris
sphincter by pilocarpine pulls the peripheral iris away from the trabecular
meshwork, thereby opening the path for aqueous outflow.
Pilocarpine is the first
choice among cholinomimet-ics for the treatment of glaucoma. Pilocarpine can be
applied to the eye as a gel (Pilopine HS Gel) or time-release system (Ocusert)
for the chronic treatment of open-angle glaucoma, or as drops (Pilocar) for an
acute reduction of intraocular pressure, as in the emergency management of
angle-closure glaucoma. Carbachol is sometimes effective in treating cases of
open-angle glaucoma that are resistant to pilocarpine.
Surgery of the Eye
Because ACh is rapidly
inactivated by cholinesterases, its use is best suited for clinical
applications requiring only a brief duration of action, such as when it is
em-ployed to cause miosis during cataract surgery. Ach (Miochol) can produce a
brief (10 minutes) miosis, and carbachol is used during eye surgery necessitating
mio-sis of a longer duration.
Urinary Retention
Bethanechol is used to treat
postsurgical bladder dys-function associated with the retention of urine. It is
most commonly given orally for this purpose, although the subcutaneous route is
also used. Effects are more rapid and intense after subcutaneous
administration, but the duration of action is shorter.
Diagnosis of Bronchial Hyperreactivity
Methacholine is used to
identify bronchial hyperreac-tivity in patients without clinically apparent
asthma. For this indication, the drug is administered by inhalation, and
patients who may be developing asthma usually produce an exaggerated airway
contraction. Upon com-pletion of the test, a rapid-acting bronchodilator (e.g.,
inhaled β-adrenoceptor agonist) can be given to counter the
bronchoconstrictor effect of methacholine and relieve the patient’s discomfort.
Adverse Effects
Potentially severe adverse
effects can result from sys-temic administration of cholinomimetic drugs, and
none should be administered by intramuscular or intra-venous injection. If
significant amounts of these drugs enter the circulation, nausea, abdominal
cramps, diar-rhea, salivation, hypotension with reflex tachycardia, cu-taneous
vasodilation, sweating, and bronchoconstric-tion can result. Pilocarpine can
cross the blood-brain barrier and affect cognitive function. Even the topical
application of cholinomimetics to the eyes can present some risk, and the
escape of cholinomimetics into the circulatory system following topical application
to the eye can be minimized by pressure applied to the lacrimal duct. Within
the eye, cholinomimetics elicit miosis and spasm of accommodation, both of
which dis-turb vision.
Bethanechol is relatively
selective in activating choli-noreceptors in the gastrointestinal and urinary
tracts when taken orally, but it is less selective when given sub-cutaneously,
and it is very dangerous when given intra-muscularly or intravenously, having
the potential to pro-duce circulatory collapse and cardiac arrest. Systemic
poisoning with cholinomimetics can be treated with the muscarinic receptor
antagonist atropine.
Bethanechol should not be
used in patients with pos-sible mechanical obstruction of the bladder or
gastroin-testinal tract or when contraction of smooth muscles in these tissues
may be harmful (e.g., recent intestinal re-section). It is also contraindicated
in patients with bronchial asthma, peptic ulcer disease, coronary artery
disease, gastrointestinal hypermotility or inflammatory disease, hypotension or
marked bradycardia, hyperthy-roidism, parkinsonism, or epilepsy. Care should be
exer-cised in administering pilocarpine to elderly patients be-cause it can
enter the CNS and affect memory and cognition, even when applied topically to
the eye.
Related Topics