Chapter: Modern Pharmacology with Clinical Applications: Directly and Indirectly Acting Cholinomimetics
Cholinesterase Inhibitors
CHOLINESTERASE
INHIBITORS
Inhibition of AChE slows or
prevents the degradation of ACh released at synapses, and this can greatly
prolong the activation of cholinoreceptors produced by synapti-cally released
ACh. In a functional sense, the indirect cholinomimetic effect of AChE
inhibitors is more selec-tive than the effect of directly acting
cholinomimetics, because the inhibitors of AChE increase the activation of
cholinoreceptors only at active cholinergic synapses. This permits
strengthening of the phasic stimulation of synaptically activated
cholinoreceptors rather than the persistent activation by directly acting
cholinomimetics. At therapeutic concentrations, inhibitors of AChE do not
activate cholinoreceptors at sites that do not receive cholinergic synaptic
input, such as endothelial mus-carinic receptors, and therefore do not present
the same risk of eliciting large vasodilator responses.
Acetylcholinesterase can be
inhibited by two gen-eral mechanisms. In the first mechanism, positively
charged quaternary ammonium compounds bind to the anionic site and prevent ACh
from binding—a simple competitive inhibition. In the second mechanism, the
agents act either as a false substrate for the cho-linesterase or directly
attack the esteratic site; in both cases they covalently modify the esteratic
site and non-competitively prevent further hydrolytic activity. Either
mechanism can be effective in preventing the hydroly- sis of ACh, but they
differ markedly in their pharmaco-kinetic properties.
Inhibition of AChE can increase
the stimulation of both muscarinic and nicotinic receptors produced by
synaptically released ACh. Nicotinic receptors can also be stimulated directly
by AChE inhibitors with a qua-ternary ammonium group, and this can potentiate
their cholinomimetic effect. Finally, although inhibition of true AChE is most
important for potentiating the synaptic activity of ACh, several AChE
inhibitors also inhibit the pseudo-ChE in plasma. This can permit plasma
concentrations of ACh to rise markedly and ac-tivate endothelial muscarinic
receptors.
Quaternary Ammonium Agents
Edrophonium (Enlon, Tensilon) and ambenonium (Mytelase) are monoquaternary and
bisquaternary am-monium alcohols, respectively. Their positive charge al-lows
them to bind to the anionic site at the reactive cen-ter, competitively
displacing ACh from the active site without covalent modification of the site.
Edrophonium has a very short duration of action, lasting only 5 to 10 minutes,
whereas inhibition by ambenonium can last 4 to 8 hours. These drugs have direct
agonist activity at nicotinic receptors.
Carbamates
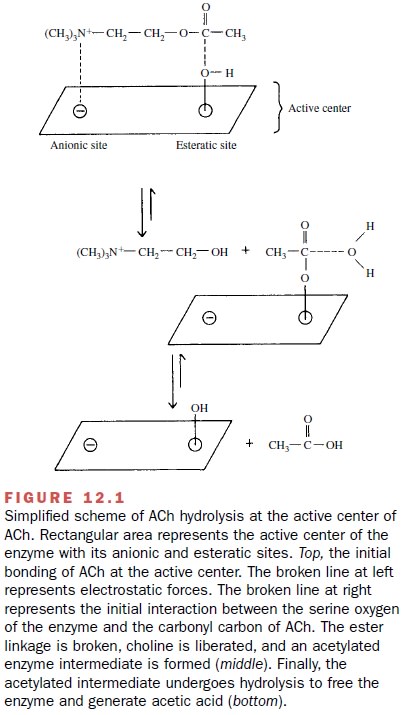
Carbamate anticholinesterase
agents are carbamic acid esters that are hydrolyzed by AChE in a manner similar
to that of ACh. Carbamates have this general structure:
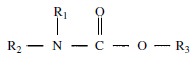
The clinically useful
carbamates generally contain a tertiary or quaternary amine group that can bind
non-covalently to the anionic site of the enzyme. The inhibi-tion of AChE by neostigmine (Prostigmin) illustrates the
general mechanism. The quaternary ammonium group of neostigmine binds
electrostatically to the an-ionic site of the enzyme, thereby orienting the
drug. The serine oxygen at the esteratic site of the enzyme then reacts with
the carbonyl carbon of neostigmine, just as it did with ACh (Fig. 12.1). However,
a carbamylated in-termediate is formed instead of an acetylated one, and this
carbamylated enzyme undergoes hydrolysis much more slowly. Whereas the
acetylated enzyme is hy-drolyzed nearly instantly, the half-life for hydrolysis
of this particular carbamylated intermediate is about an hour. The carbamates
generally inhibit pseudo-ChE as well as true AChE, and their suicidal
degradation by cholinesterases contributes importantly to terminating their
duration of effect. Physostigmine (also called eser-ine) (Antilirium) is a tertiary amine that can inhibit AChE in the CNS,
and it can be used in life-threatening cases to treat antimuscarinic poisoning.
Pyridostigmine (Mestinon) is a quaternary ammo-nium
carbamate. Neostigmine and pyridostigmine also have direct agonist activity at
nicotinic receptors on skeletal muscle. Rivastigmine (Exelon) is a carbamate cholinesterase inhibitor with good
penetration into the brain.
Organophosphates
The organophosphate compounds
also react at the es-teratic site of AChE (Fig. 12.4). In general, however,
they are much less selective than are the carbamates, in-hibiting many enzymes
that contain a serine molecule at an active center. The organophosphate
compounds have this general structure:
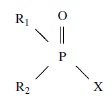
Examples of X groups are
fluorine in isoflurophate (Floropryl,
no longer available in the United States or Canada) and echothiophate (Phospholine). Parathion and malathion
(insecticides) are thiophosphates

that must be converted to
oxyanalogues to become ac-tive. The organophosphates, except for echothiophate,
are very lipid soluble.
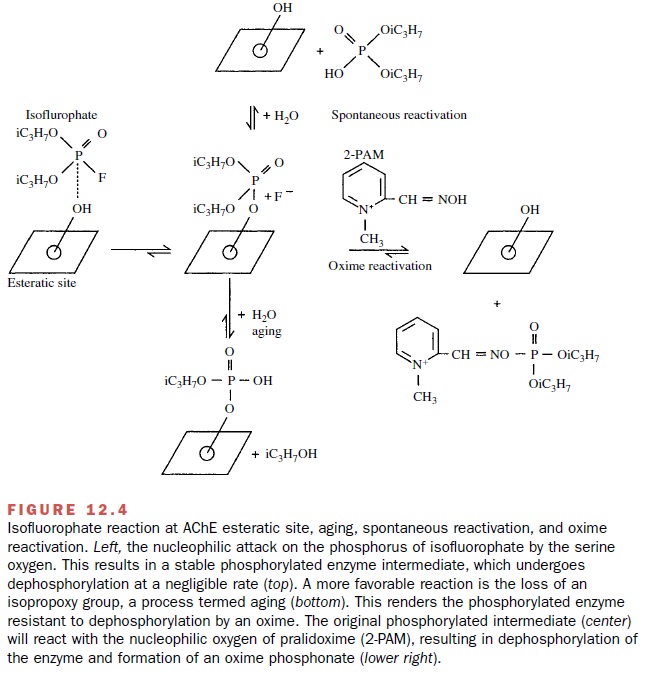
In the interaction of isoflurophate with AChE, a phosphorylated intermediate is formed and fluoride is released.An important characteristic of the organophos-phate-induced inhibition is that the bond between the phosphate and the enzyme is very stable. While the re-generation of most carbamylated enzymes occurs with a half-life of minutes or hours, the recovery of a phospho-rylated enzyme is generally measured in days. These agents are referred to, therefore, as irreversible in-hibitors.
Although the spontaneous
hydrolysis of a phospho-rylated enzyme is generally very slow, compounds called
oximes can cause dephosphorylation (Fig. 12.4). Pralidoxime chloride (2-PAM)
(Protopam chloride) is an oxime used therapeutically to reactivate
phosphory-lated AChE. It has the additional feature that its qua-ternary
ammonium group binds to the anionic site of the enzyme and thereby promotes
dephosphorylation. If the oxime is not administered soon enough (minutes to
hours) after AChE has been inhibited, an alkoxy group may be lost from the
phosphorylated enzyme. This reaction is called aging. Once aging has occurred,
oximes can no longer regenerate free enzymes. The rate of aging appears to
depend both on the nature of the enzyme (AChE or pseudo-ChE) and on the
particular inhibitor employed. Since pralidoxime is a quaternary amine, it does
not cross the blood-brain barrier, and it is not useful for reactivating
cholinesterases in the CNS.
Inhibitors targeted at AChE in the CNS
Several inhibitors of AChE
have been developed for use in treating Alzheimer’s disease, which requires
that the drugs readily enter the CNS. These inhibitors are struc-turally
unrelated and vary in their mechanism of inhibi-tion, although all are
reversible inhibitors. Tacrine (Cognex)
is a monoamine acridine. Donepezil (Aricept)
is a piperidine derivative that is a relatively specific in-hibitor of AChE in
the brain, with little effect on pseudo-ChE in the periphery. Galanthamine (Reminyl ) is a ter-tiary alkaloid and
phenanthrene derivative extracted from daffodil bulbs that is a reversible
competitive in-hibitor of AChE; it also acts on nicotinic receptors.
Absorption, Metabolism, and Excretion
Physostigmine and
rivastigmine are tertiary amines that are rapidly absorbed from the
gastrointestinal tract, as are tacrine, donepezil, and galanthamine, whereas
qua-ternary ammonium compounds are poorly absorbed after oral administration.
Nevertheless, quaternary am-monium compounds like neostigmine and
pyridostig-mine are orally active if larger doses are employed. Only the
quaternary ammonium inhibitors do not read-ily enter the CNS. Because of their
high lipid solubility and low molecular weight, most of the organophos-phates
are absorbed by all routes of administration; even percutaneous exposure can
result in the absorp-tion of sufficient drug to permit the accumulation of
toxic levels of these compounds.
Edrophonium is partially
metabolized to a glu-curonide conjugate in the liver. Some of this metabolite
is excreted in bile. Carbamates undergo both nonenzy-matic and enzymatic
hydrolysis, with enzymatic hydrol-ysis generally resulting from an interaction
of the drug with the pseudo-ChE in plasma and liver. Organo- phosphates are
metabolized to inactive products by hy-drolytic enzymes in the plasma, kidney,
liver, and lungs. In contrast, the organophosphate insecticide parathion
requires metabolism (oxidative desulfuration) to be-come an effective
insecticide.
Metabolites of the
cholinesterase inhibitors and in some instances significant amounts of the
parent com-pound are eliminated in the urine. Renal excretion is very important
in the clearance of agents such as neostigmine, pyridostigmine, and
edrophonium. This is demonstrated by a twofold to threefold increase in
elimination half-lives for these drugs in anephric pa-tients. Renal elimination
is largely the result of glomerular filtration but probably also involves, at
least in the case of quaternary amines, secretion via the renal cationic
transport system.
Basic Pharmacology
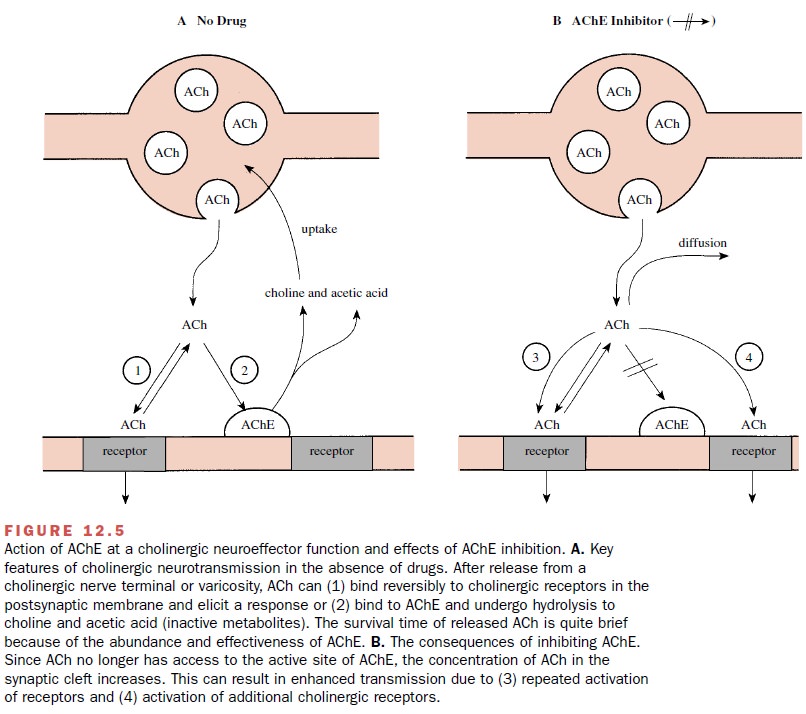
Inhibition of AChE
potentiates and prolongs the stimu-lation of cholinoreceptors resulting from
ACh released at cholinergic synapses (Fig. 12.5). These synapses in-clude those
found at the skeletal neuromuscular junc-tion, adrenal medulla, autonomic
ganglia, cholinergic neuroeffector junctions of the autonomic nervous sys-tem,
and cholinergic synapses in the CNS. The degree and range of effects observed
depend on the inhibitor chosen, the dose employed, and the route of exposure or
administration.
Neuromuscular transmission in
skeletal muscle is enhanced by low concentrations of anticholinesterase agents,
whereas high concentrations result in choliner-gic blockade. This blockade is
initially due to a persis-tent membrane depolarization and inactivation of
voltage-gated sodium channels, but if ACh levels remain high, the nicotinic
cholinergic receptors can quickly be-come desensitized. Although
anticholinesterase agents will facilitate cholinergic transmission at autonomic
gan-glia, their action at these sites is less marked than at the neuromuscular
junction.
Anticholinesterase agents of
all classes can initiate antidromic firing of action potentials in motor
neurons, possibly due to an activation of prejunctional ACh re-ceptors that are
activated by the elevated synaptic ACh. Quaternary ammonium inhibitors can also
act as ago-nists at these receptors. The initiation of antidromic fir-ing may
be a mechanism by which cholinesterase in-hibitors produce fasciculation of
skeletal muscle.
The actions of anticholinesterase agents on the car-diovascular system are complex. The primary effect pro-duced by potentiation of vagal stimulation is brady-cardia with a consequent decrease in cardiac output and blood pressure.
However, potentiation of both parasympathetic and sympathetic ganglionic transmission, including that in the
adrenal medulla, can produce complicated effects on the cardiovascular system,
in-cluding vasoconstrictor responses. The activation of re-flexes can also
complicate the total cardiovascular re-sponse to cholinesterase inhibitors.
Related Topics