Chapter: Basic & Clinical Pharmacology : Cholinoceptor Blocking Drugs
Clinical Pharmacology of the Muscarinic Receptor Blocking Drugs
CLINICAL PHARMACOLOGY OF THE MUSCARINIC RECEPTOR BLOCKING DRUGS
Therapeutic Applications
The antimuscarinic drugs have applications in several of the major organ systems and in the treatment of poisoning by muscarinic agonists.
A. Central Nervous System Disorders
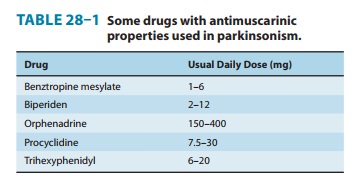
· Parkinson’s disease—The treatment of Parkinson’s diseaseis often an exercise in polypharmacy, since no single agent is fully effective over the course of the disease. Most antimuscarinic drugs promoted for this application (see Table 28–1) were developed before levodopa became available. Their use is accompanied by all of the adverse effects described below, but the drugs remain usefulas adjunctive therapy in some patients.
· Motion sickness—Certain vestibular disorders respond toantimuscarinic drugs (and to antihistaminic agents with antimus-carinic effects). Scopolamine is one of the oldest remedies for seasickness and is as effective as any more recently introduced agent. It can be given by injection or by mouth or as a transdermal patch. The patch formulation produces significant blood levels over 48–72 hours. Useful doses by any route usually cause signifi-cant sedation and dry mouth.
B. Ophthalmologic Disorders
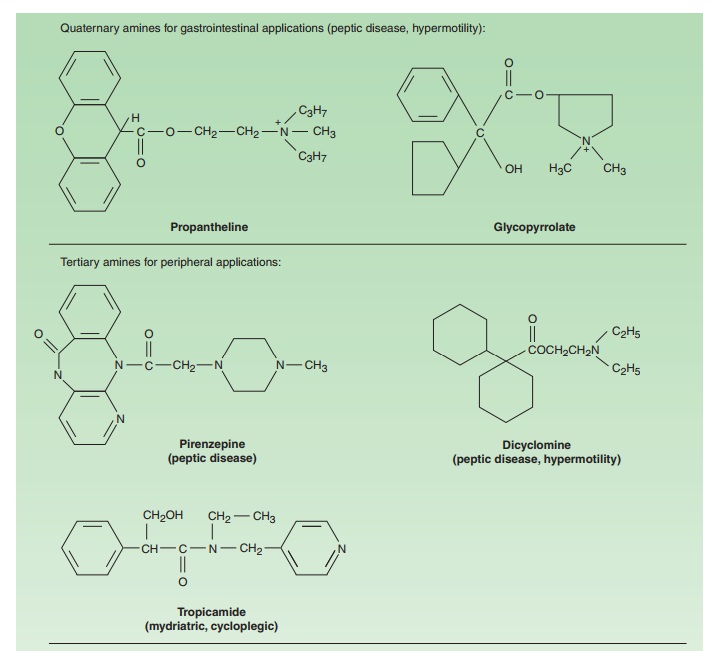
Accurate measurement of refractive error in uncooperative patients, eg, young children, requires ciliary paralysis. Also, ophthalmo-scopic examination of the retina is greatly facilitated by mydriasis. Therefore, antimuscarinic agents, administered topically as eye drops or ointment, are very helpful in doing a complete examina-tion. For adults and older children, the shorter-acting drugs are preferred (Table 8–2). For younger children, the greater efficacy of atropine is sometimes necessary, but the possibility of antimusca-rinic poisoning is correspondingly increased. Drug loss from the conjunctival sac via the nasolacrimal duct into the nasopharynx can be diminished by the use of the ointment form rather than drops. Formerly, ophthalmic antimuscarinic drugs were selected from the tertiary amine subgroup to ensure good penetration after conjunctival application. Recent experiments in animals, however, suggest that glycopyrrolate, a quaternary agent, is as rapid in onset and as long-lasting as atropine.
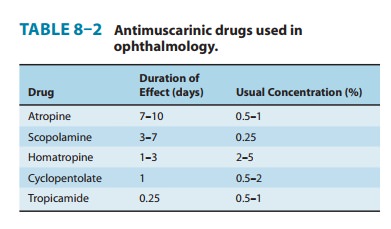
Antimuscarinic drugs should never be used for mydriasis unless cycloplegia or prolonged action is required. Alpha-adrenoceptor stimulant drugs, eg, phenylephrine, produce a short-lasting mydriasis that is usually sufficient for funduscopic examination .A second ophthalmologic use is to prevent synechia (adhesion) formation in uveitis and iritis. The longer-lasting preparations, especially homatropine, are valuable for this indication.
C. Respiratory Disorders
The use of atropine became part of routine preoperative medica-tion when anesthetics such as ether were used, because these irri-tant anesthetics markedly increased airway secretions and were associated with frequent episodes of laryngospasm. Preanesthetic injection of atropine or scopolamine could prevent these hazard-ous effects. Scopolamine also produces significant amnesia for the events associated with surgery and obstetric delivery, a side effect that was considered desirable. On the other hand, urinary reten-tion and intestinal hypomotility following surgery were often exacerbated by antimuscarinic drugs. Newer inhalational anesthet-ics are far less irritating to the airways.
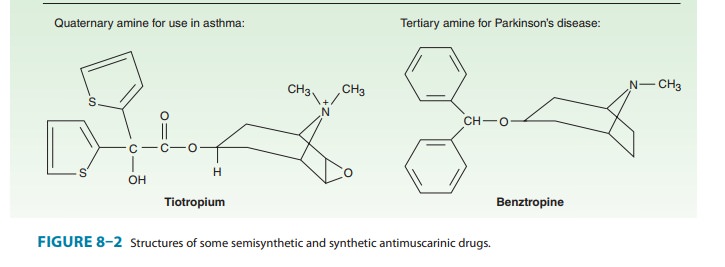
Patients with COPD, a condition that occurs more frequently in older patients, particularly chronic smokers, benefit from broncho-dilators, especially antimuscarinic agents. Ipratropium and tiotro-pium (see Figure 8–2), synthetic analogs of atropine, are used asinhalational drugs in COPD. The aerosol route of administration has the advantage of maximal concentration at the bronchial target tissue with reduced systemic effects. This application is discussed in greater detail. Tiotropium has a longer bronchodila-tor action than ipratropium and can be given once daily because it dissociates slowly from M3 receptors. It has a terminal elimination t1/2of 5–6 days; steady-state plasma levels are achieved in about 25days with single daily administration. Tiotropium reduces the inci-dence of COPD exacerbations and is a useful adjunct to pulmonary rehabilitation in increasing exercise tolerance. The hyperactive neu-ral bronchoconstrictor reflex present in most individuals with asthma is mediated by the vagus, acting on muscarinic receptors onbronchial smooth muscle cells. Ipratropium and tiotropium are also used as inhalational drugs in asthma.
D. Cardiovascular Disorders
Marked reflex vagal discharge sometimes accompanies the pain of myocardial infarction (eg, vasovagal attack) and may depress sinoatrial or atrioventricular node function sufficiently to impair cardiac output. Parenteral atropine or a similar antimuscarinic drug is appropriate therapy in this situation. Rare individuals without other detectable cardiac disease have hyperactive carotid sinus reflexes and may experience faintness or even syncope as a result of vagal discharge in response to pressure on the neck, eg, from a tight collar. Such individuals may benefit from the judi-cious use of atropine or a related antimuscarinic agent.
Pathophysiology can influence muscarinic activity in other ways as well. Circulating autoantibodies against the second extracellular loop of cardiac M2 muscarinic receptors have been detected in some patients with idiopathic dilated cardiomyopathy and those afflicted with Chagas’ disease caused by the protozoan Trypanosomacruzi. Patients with Graves’ disease (hyperthyroidism) also havesuch autoantibodies that may facilitate the development of atrial fibrillation. These antibodies exert parasympathomimetic actions on the heart that are prevented by atropine. In animals immunized with a peptide from the second extracellular loop of the M2 recep-tor, the antibody is an allosteric modulator of the receptor. Although their role in the pathology of heart diseases is unknown, these antibodies should provide clues to the molecular basis of receptor activation because their site of action differs from the orthosteric site where acetylcholine binds .
E. Gastrointestinal Disorders
Antimuscarinic agents are now rarely used for peptic ulcer disease in the USA . Antimuscarinic agents can provide some relief in the treatment of common traveler’s diarrhea and other mild or self-limited conditions of hypermotility. They are often combined with an opioid antidiarrheal drug, an extremely effective therapy. In this combination, however, the very low dos-age of the antimuscarinic drug functions primarily to discourage abuse of the opioid agent. The classic combination of atropine with diphenoxylate, a nonanalgesic congener of meperidine, is available under many names (eg, Lomotil) in both tablet and liq-uid form .
F. Urinary Disorders
Atropine and other antimuscarinic drugs have been used to provide symptomatic relief in the treatment of urinary urgency caused by minor inflammatory bladder disorders (Table 8–3). However, spe-cific antimicrobial therapy is essential in bacterial cystitis. In the human urinary bladder, M2 and M3 receptors are expressed pre-dominantly with the M3 subtype mediating direct activation of contraction. As in intestinal smooth muscle, the M2 subtype appears to act indirectly by inhibiting relaxation by norepinephrine and epinephrine.
Receptors for acetylcholine on the urothelium (the epithelial lining of the urinary tract) and on afferent nerves as well as the detrusor muscle provide a broad basis for the action of antimusca-rinic drugs in the treatment of overactive bladder.Oxybutynin,
It is also valuable in reducing involuntary voiding in patients with neuro-logic disease, eg, children with meningomyelocele. Oral oxybu-tynin or instillation of the drug by catheter into the bladder in such patients appears to improve bladder capacity and continence and to reduce infection and renal damage. Transdermally applied oxy-butynin or its oral extended-release formulation reduces the need for multiple daily doses. Trospium, a nonselective antagonist, has been approved and is comparable in efficacy and side effects to oxybutynin. Darifenacin and solifenacin are recently approved antagonists that have greater selectivity for M3 receptors than oxy-butynin or trospium. Darifenacin and solifenacin have the advan-tage of once-daily dosing because of their long half-lives. Tolterodine and fesoterodine,M3-selective antimuscarinics, are available for use in adults with urinary incontinence. They have many of the quali-ties of darifenacin and solifenacin and are available in extended-release tablets. The convenience of the newer and longer-acting drugs has not been accompanied by improvements in overall effi-cacy or by reductions in side effects such as dry mouth. An alterna-tive treatment for urinary incontinence refractory to antimuscarinic drugs is intrabladder injection of botulinum toxin A. Botulinum toxin is reported to reduce urinary incontinence for several months after a single treatment by interfering with the co-release of ATP with neuronal acetylcholine (see Figure 6–3). Blockade of the acti-vation of sensory nerves in the urothelium by ATP may account for a large part of this effect of botulinum toxin. This approach is not an FDA-approved indication at present.
Imipramine, a tricyclic antidepressant drug with strong anti-muscarinic actions, has long been used to reduce incontinence in institutionalized elderly patients. It is moderately effective but causes significant CNS toxicity.Propiverine, a newer antimusca-rinic agent, has been approved for this purpose.
Antimuscarinic agents have also been used in urolithiasis to relieve the painful ureteral smooth muscle spasm caused by pas-sage of the stone. However, their usefulness in this condition is debatable.
G. Cholinergic Poisoning
Severe cholinergic excess is a medical emergency, especially in rural communities where cholinesterase inhibitor insecticides are com-monly used and in cultures where wild mushrooms are frequently eaten. The potential use of cholinesterase inhibitors as chemical warfare “nerve gases” also requires an awareness of the methods for treating acute poisoning .
1. Antimuscarinic therapy— Both the nicotinic and the mus-carinic effects of the cholinesterase inhibitors can be life-threatening. Unfortunately, there is no effective method for directly blocking the nicotinic effects of cholinesterase inhibition, because nicotinic agonists and antagonists cause blockade of transmission . To reverse the muscarinic effects, a tertiary (not quaternary) amine drug must be used (preferably atropine) to treat the CNS effects as well as the peripheral effects of the organophos-phate inhibitors. Large doses of atropine may be needed to oppose the muscarinic effects of extremely potent agents like parathion and chemical warfare nerve gases: 1–2 mg of atropine sulfate may be given intravenously every 5–15 minutes until signs of effect (dry mouth, reversal of miosis) appear. The drug may have to be given many times, since the acute effects of the cholinesterase inhibitor may last 24–48 hours or longer. In this life-threatening situation, as much as 1 g of atropine per day may be required for as long as 1 month for full control of muscarinic excess.
2. Cholinesterase regenerator compounds—A second classof compounds, composed of substituted oximes capable of regen-erating active enzyme from the organophosphorus-cholinesterase complex, is also available to treat organophosphorus poisoning. These oxime agents include pralidoxime (PAM), diacetylmon-oxime (DAM), obidoxime, and others.
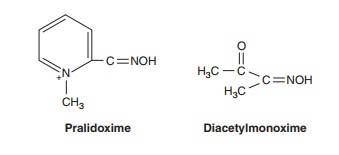
Organophosphates cause phosphorylation of the serine OH group at the active site of cholinesterase. The oxime group (ÓNOH) has a very high affinity for the phosphorus atom, for which it competes with serine OH. These oximes can hydrolyze the phosphorylated enzyme and regenerate active enzyme from the organophosphorus-cholinesterase complex if the complex has not “aged” . Pralidoxime is the most extensively studied—in humans—of the agents shown and the only one avail-able for clinical use in the USA. It is most effective in regenerating the cholinesterase associated with skeletal muscle neuromuscular junctions. Pralidoxime and obidoxime are ineffective in reversing the central effects of organophosphate poisoning because each has positively charged quaternary ammonium groups that prevent entry into the CNS. Diacetylmonoxime, on the other hand, crosses the blood-brain barrier and, in experimental animals, can regenerate some of the CNS cholinesterase.
Pralidoxime is administered by intravenous infusion, 1–2 g given over 15–30 minutes. In spite of the likelihood of aging of the phosphate-enzyme complex, recent reports suggest that administration of multiple doses of pralidoxime over several days may be useful in severe poisoning. In excessive doses, pralidoxime can induce neuromuscular weakness and other adverse effects.
A third approach to protection against excessive acetylcholin-esterase inhibition is pretreatment with reversible enzyme inhibi-tors to prevent binding of the irreversible organophosphate inhibitor. This prophylaxis can be achieved with pyridostigmine but is reserved for situations in which possibly lethal poisoning is anticipated, eg, chemical warfare . Simultaneous use of atropine is required to control muscarinic excess.
Mushroom poisoning has traditionally been divided intorapid-onset and delayed-onset types. The rapid-onset type is usu-ally apparent within 30 minutes to 2 hours after ingestion of the mushrooms, and can be caused by a variety of toxins. Some of these produce simple upset stomach; others can have disulfiram-like effects; some cause hallucinations; and a few mushrooms (eg, Inocybe species) can produce signs of muscarinic excess: nausea, vomiting, diarrhea, urinary urgency, sweating, salivation, and sometimes bronchoconstriction. Parenteral atropine, 1–2 mg, is effective treatment in such intoxications. Despite its name, Amanita muscaria contains not only muscarine (the alkaloid wasnamed after the mushroom), but also numerous other alkaloids, including antimuscarinic agents, and ingestion of A muscaria often causes signs of atropine poisoning, not muscarine excess.
Delayed-onset mushroom poisoning, usually caused by Amanita phalloides, A virosa, Galerina autumnalis, or G marginata, manifests its first symptoms 6–12 hours after ingestion. Although the initial symptoms usually include nausea and vomiting, the major toxicity involves hepatic and renal cellular injury by ama-toxins that inhibit RNA polymerase. Atropine is of no value in this form of mushroom poisoning .
H. Other Applications
Hyperhidrosis (excessive sweating) is sometimes reduced by anti-muscarinic agents. However, relief is incomplete at best, probably because apocrine rather than eccrine glands are usually involved.
Adverse Effects
Treatment with atropine or its congeners directed at one organ system almost always induces undesirable effects in other organ systems. Thus, mydriasis and cycloplegia are adverse effects when an antimuscarinic agent is used to reduce gastrointestinal secretion or motility, even though they are therapeutic effects when the drug is used in ophthalmology.
At higher concentrations, atropine causes block of all parasym-pathetic functions. However, atropine is a remarkably safe drug inadults. Atropine poisoning has occurred as a result of attemptedsuicide, but most cases are due to attempts to induce hallucina-tions. Poisoned individuals manifest dry mouth, mydriasis, tachy-cardia, hot and flushed skin, agitation, and delirium for as long as 1 week. Body temperature is frequently elevated. These effects are memorialized in the adage, “dry as a bone, blind as a bat, red as a beet, mad as a hatter.”
Unfortunately, children, especially infants, are very sensitive to the hyperthermic effects of atropine. Although accidental admin-istration of over 400 mg has been followed by recovery, deaths have followed doses as small as 2 mg. Therefore, atropine should be considered a highly dangerous drug when overdose occurs in infants or children.
Overdoses of atropine or its congeners are generally treated symptomatically . Poison control experts discour-age the use of physostigmine or another cholinesterase inhibitor to reverse the effects of atropine overdose because symptomatic man-agement is more effective and less dangerous. When physostig-mine is deemed necessary, smalldoses are given slowly intravenously (1–4 mg in adults, 0.5–1 mg in children). Symptomatic treatment may require temperature control with cooling blankets and seizure control with diazepam.
Poisoning caused by high doses of quaternary antimuscarinic drugs is associated with all of the peripheral signs of parasympathetic blockade but few or none of the CNS effects of atropine. These more polar drugs may cause significant ganglionic blockade, how-ever, with marked orthostatic hypotension . Treatment of the antimuscarinic effects, if required, can be carried out with a quaternary cholinesterase inhibitor such as neostigmine. Control of hypotension may require the administration of a sympathomi-metic drug such as phenylephrine.
Recent evidence indicates that some centrally acting drugs (tricyclic antidepressants, selective serotonin reuptake inhibitors, anti-anxiety agents) with antimuscarinic actions impair memory and cognition in older patients.
Contraindications
Contraindications to the use of antimuscarinic drugs are relative, not absolute. Obvious muscarinic excess, especially that caused by cholinesterase inhibitors, can always be treated with atropine.Antimuscarinic drugs are contraindicated in patients with glaucoma, especially angle-closure glaucoma. Even systemic use of moderate doses may precipitate angle closure (and acute glau-coma) in patients with shallow anterior chambers.In elderly men, antimuscarinic drugs should always be used with caution and should be avoided in those with a history of prostatic hyperplasia.
Because the antimuscarinic drugs slow gastric emptying, they may increase symptoms in patients with gastric ulcer. Nonselective antimuscarinic agents should never be used to treat acid-peptic disease .
Related Topics