Chapter: Basic & Clinical Pharmacology : Antipsychotic Agents & Lithium
Basic Pharmacology of Lithium
BASIC PHARMACOLOGY OF LITHIUM
Lithium was first used
therapeutically in the mid-19th century in patients with gout. It was briefly
used as a substitute for sodium chloride in hypertensive patients in the 1940s
but was banned after it proved too toxic for use without monitoring. In 1949,
Cade dis-covered that lithium was an effective treatment for bipolar disorder,
engendering a series of controlled trials that confirmed its efficacy as
monotherapy for the manic phase of bipolar disorder.
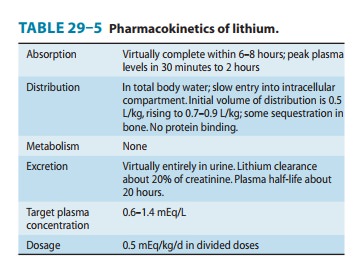
Pharmacokinetics
Lithium is a small monovalent
cation. Its pharmacokinetics are summarized in Table 29–5.
Pharmacodynamics
Despite considerable investigation, the biochemical basis for mood stabilizer therapies including lithium and anticonvulsant mood stabilizers is not clearly understood. Lithium directly inhibits two signal transduction pathways.
It both suppresses inositol signaling through depletion of
intracellular inositol and inhibits glycogen synthase kinase-3 (GSK-3), a
multifunctional protein kinase. GSK-3 is a component of diverse intracellular
signaling pathways. These include signaling via insulin/insulin-like growth
factor, brain-derived neurotrophic factor (BDNF), and the Wnt pathway. All of
these lead to inhibition of GSK-3. GSK-3 phosphorylates β-catenin, resulting in
interaction with transcription factors. The pathways that are facilitated in
this manner modulate energy metabolism, provide neuroprotection, and increase
neuroplasticity.
Studies on the enzyme
prolyl oligopeptidase and the sodium myoinositol transporter support an
inositol depletion mechanism for mood-stabilizer action. Valproic acid may
indirectly reduce GSK-3 activity and can up-regulate gene expression through
inhi-bition of histone deacetylase. Valproic acid also inhibits inositol
signaling through an inositol depletion mechanism. There is no evidence of
GSK-3 inhibition by carbamazepine, a second antiepi-leptic mood stabilizer. In
contrast, this drug alters neuronal mor-phology through an inositol depletion
mechanism, as seen with lithium and valproic acid. The mood stabilizers may
also have indirect effects on neurotransmitters and their release.
A. Effects on Electrolytes and
Ion Transport
Lithium is closely
related to sodium in its properties. It can substitute for sodium in generating
action potentials and in Na+-Na+ exchange across the
membrane. It inhibits the latter process; that is, Li+-Na+
exchange is gradually slowed after lithium is introduced into the body. At
therapeutic concentrations (around 1 mmol/L), it does not signifi-cantly affect
the Na+-Ca2+ exchanger or the Na+/K+-ATPase
pump.
B. Effects on Second Messengers
Some of the enzymes
affected by lithium are listed in Table 29–6. One of the best-defined effects
of lithium is its action on inositol

Early
studies of lithium demonstrated changes in brain inositol phosphate levels, but
the significance of these changes was not appreciated until the
second-messenger roles of inositol-1,4,5-trisphosphate (IP3) and diacylglycerol
(DAG) were discovered. As described, inositol trisphosphate and diacylglycerol
are important second messengers for both α-adrenergic and muscarinic transmission.
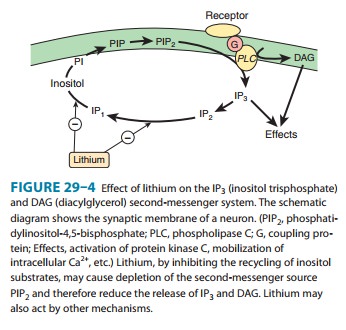
Lithium inhibits inositol monophosphatase (IMPase) and other important enzymes
in the normal recycling of membrane phosphoinositides, including conversion of
IP2 (inositol
diphos-phate) to IP1 (inositol monophosphate) and the conversion of IP1 to inositol (Figure
29–4). This block leads to a depletion of free inositol and ultimately of
phosphatidylinositol-4,5-bisphosphate (PIP2), the membrane precursor of IP3 and DAG. Over time,
the effects of transmitters on the cell diminish in proportion to the amount of
activity in the PIP2-dependent pathways. The activity of these pathways is
postulated to be markedly increased during a manic episode. Treatment with
lithium would be expected to diminish the activity in these circuits.
Studies of
noradrenergic effects in isolated brain tissue indicate that lithium can
inhibit norepinephrine-sensitive adenylyl cyclase. Such an effect could relate
to both its antidepressant and its anti-manic effects. The relationship of
these effects to lithium’s actions on IP3 mechanisms is currently unknown.
Because lithium
affects second-messenger systems involving both activation of adenylyl cyclase
and phosphoinositol turnover, it is not surprising that G proteins are also
found to be affected. Several studies suggest that lithium may uncouple
receptors from their G proteins; indeed, two of lithium’s most common side
effects, polyuria and subclinical hypothyroidism, may be due to uncoupling of
the vasopressin and thyroid-stimulating hormone (TSH) receptors from their G
proteins.
The major current
working hypothesis for lithium’s therapeutic mechanism of action supposes that
its effects on phosphoinositolturnover, leading to an early relative reduction
of myoinositol in human brain, are part of an initiating cascade of
intracellular changes. Effects on specific isoforms of protein kinase C may be
most relevant. Alterations of protein kinase C-mediated signaling alter gene
expres-sion and the production of proteins implicated in long-term neuro-plastic
events that could underlie long-term mood stabilization.
Related Topics