Chapter: Basic & Clinical Pharmacology : Antipsychotic Agents & Lithium
Basic Pharmacology of Antipsychotic Agents
BASIC PHARMACOLOGY OF ANTIPSYCHOTIC AGENTS
Chemical Types
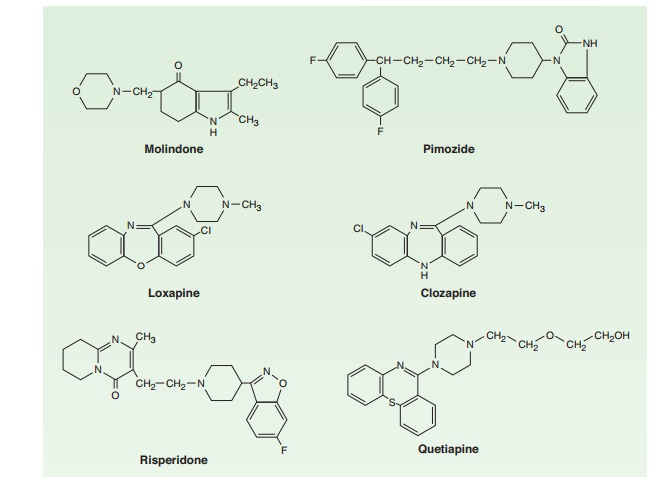
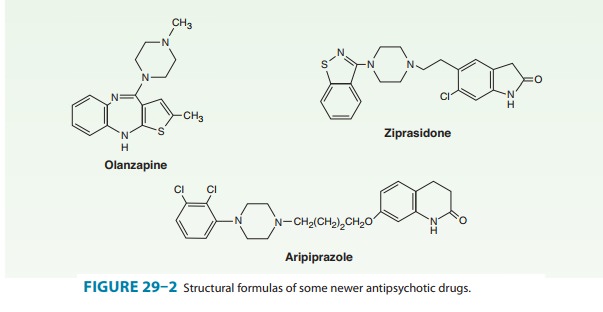
A number of chemical structures have been associated with antipsychotic properties. The drugs can be classified into several groups as shown in Figures 29–1 and 29–2.
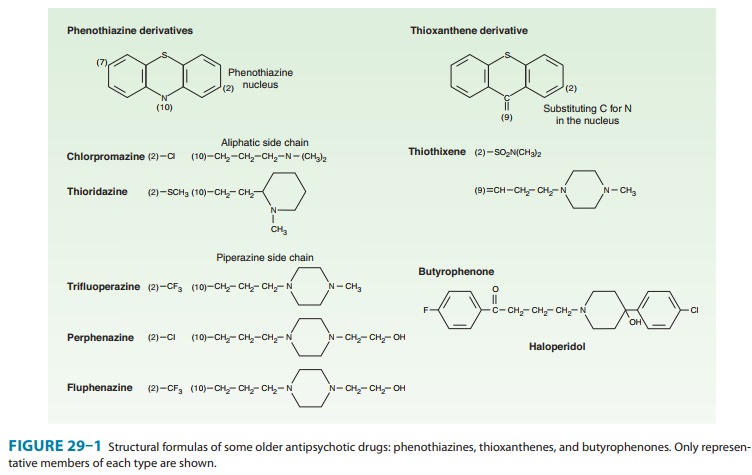
A. Phenothiazine Derivatives
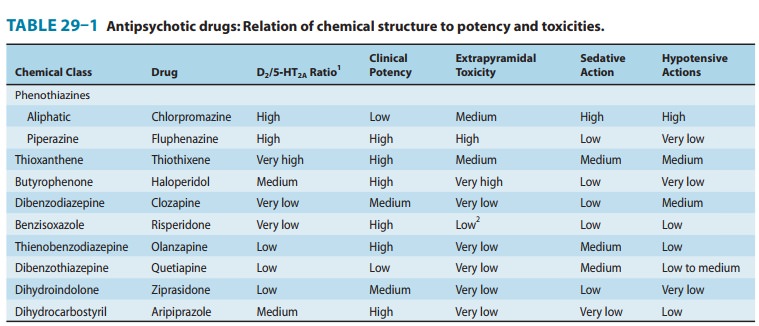
Three subfamilies of phenothiazines, based primarily on the side chain of the molecule, were once the most widely used of the antipsychotic agents. Aliphatic derivatives (eg, chlorprom-azine) and piperidine derivatives (eg, thioridazine) are theleast potent. These drugs produce more sedation and weight gain. Piperazine derivatives are more potent (effective in lower doses) but not necessarily more efficacious. Perphenazine, a piperazine derivative, was the typical antipsychotic drug used in the CATIE study described in the following text. The piperazine derivatives are also more selective in their pharma-cologic effects (Table 29–1).
Recently, a large study in the USA (CATIE) reported that per-phenazine was as effective as atypical antipsychotic drugs, with the modest exception of olanzapine, and concluded that typical antipsychotic drugs are the treatment of choice for schizophrenia based on their lower cost. However, there were numerous flaws in the design, execution and analysis of this study, leading to it having only modest impact on clinical practice. In particular, it failed to consider issues such as dosage of olanzapine, inclusion of treatment resistant patients, encouragement of patients to switch medica-tions inherent in the design, risk for tardive dyskinesia following long term use of even low dose typical antipsychotics, and the necessity of large sample sizes in equivalency studies.
B. Thioxanthene Derivatives
This group of drugs is exemplified primarily by thiothixene.
C. Butyrophenone Derivatives
This group, of which haloperidol is the most widely used, has a very different structure from those of the two preceding groups. Haloperidol, a butyrophenone, is the most widely used typical antipsychotic drug, despite its high level of EPS relative to typical antipsychotic drugs. Diphenylbutylpiperidines are closely related compounds. The butyrophenones and congeners tend to be more potent and to have fewer autonomic effects but greater extrapyra-midal effects than phenothiazines (Table 29–1).
D. Miscellaneous Structures
Pimozide and molindone are typical antipsychotic drugs. There is no significant difference in efficacy between these newer typical and the older typical antipsychotic drugs.
E. Atypical Antipsychotic Drugs
Clozapine, asenapine, olanzapine, quetiapine, paliperidone, risperidone, sertindole, ziprasidone, zotepine, andaripipra-zole are atypical antipsychotic drugs (Figure 29–2). Clozapine isthe prototype. Paliperidone is 9-hydroxyrisperidone, the active metabolite of risperidone. Risperidone is rapidly converted to 9-hydroxyrisperidone in vivo in most patients, except for about 10% of patients who are poor metabolizers. Sertindole is approved in some European countries but not in the USA.
These drugs have complex pharmacology but they share a greater ability to alter 5-HT2A-receptor activity than to interfere with D2-receptor action. In most cases, they act as partial agonistsat the 5-HT1A receptor, which produces synegistic effects with 5-HT2A receptor antagonism. Most are either 5-HT6 or 5-HT7 receptor antagonists.
Sulpride and sulpiride constitute another class of atypical agents. They have equivalent potency for D2 and D3 receptors, but they are also 5-HT7 antagonists. They dissociate EPS and antipsy-chotic efficacy. However, they also produce marked increases in serum prolactin levels and are not as free of the risk of tardive dyskinesia as are drugs such as clozapine and quetiapine.
Pharmacokinetics
A. Absorption and Distribution
Most antipsychotic drugs are readily but incompletely absorbed. Furthermore, many undergo significant first-pass metabolism. Thus, oral doses of chlorpromazine and thioridazine have sys-temic availability of 25–35%, whereas haloperidol, which has less first-pass metabolism, has an average systemic availability of about 65%.
Most antipsychotic drugs are highly lipid soluble and protein bound (92–99%). They tend to have large volumes of distribu-tion (usually more than 7 L/kg). They generally have a much longer clinical duration of action than would be estimated from their plasma half-lives. This is paralleled by prolonged occu-pancy of D2 dopamine receptors in the brain by the typical antipsychotic drugs.
Metabolites of chlorpromazine may be excreted in the urine weeks after the last dose of chronically administered drug. Long-acting injectable formulations may cause some blockade of D2 receptors 3–6 months after the last injection. Time to recurrence of psychotic symptoms is highly variable after discontinuation of antipsychotic drugs. The average time for relapse in stable patients with schizophrenia who discontinue their medication is 6 months. Clozapine is an exception in that relapse after discontinuation is usually rapid and severe. Thus, clozapine should never be discon-tinued abruptly unless clinically needed because of adverse effects such as myocarditis or agranulocytosis, which are true medical emergencies.
B. Metabolism
Most antipsychotic drugs are almost completely metabolized by oxidation or demethylation, catalyzed by liver microsomal cyto-chrome P450 enzymes. CYP2D6, CYP1A2, and CYP3A4 are the major isoforms involved . Drug-drug interac-tions should be considered when combining antipsychotic drugs with various other psychotropic drugs or drugs—such as ketoconazole—that inhibit various cytochrome P450 enzymes. At the typical clinical doses, antipsychotic drugs do not usually interfere with the metabolism of other drugs.
Pharmacodynamics
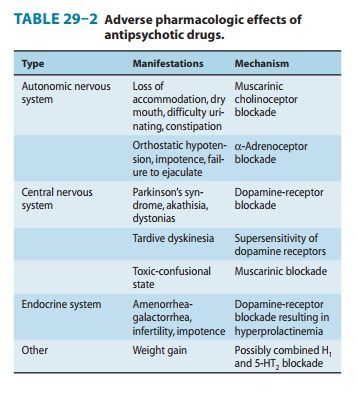
The first phenothiazine antipsychotic drugs, with chlorpromazine as the prototype, proved to have a wide variety of central nervous system, autonomic, and endocrine effects. Although efficacy of these drugs is primarily driven by D2-receptor blockade, their adverse actions were traced to blocking effects at a wide range of receptors including α adrenoceptors and muscarinic, H1 hista-minic, and 5-HT2 receptors.
A. Dopaminergic Systems
Five dopaminergic systems or pathways are important for under-standing schizophrenia and the mechanism of action of antipsy-chotic drugs. The first pathway—the one most closely related to behavior and psychosis—is the mesolimbic-mesocortical pathway, which projects from cell bodies in the ventral tegmentum in separate bundles of axons to the limbic system and neocortex. The second system—the nigrostriatal pathway—consists of neurons that project from the substantia nigra to the dorsal striatum, which includes the caudate and putamen; it is involved in the coordination of voluntary movement. Blockade of the D2 receptors in the nigrostriatal pathway is responsible for EPS. The third pathway—the tuberoinfundibular system—arises in the arcuate nuclei and periventricular neurons and releases dop-amine into the pituitary portal circulation. Dopamine released by these neurons physiologically inhibits prolactin secretion from the anterior pituitary. The fourth dopaminergic system— the medullary-periventricular pathway—consists of neurons in the motor nucleus of the vagus whose projections are not well defined. This system may be involved in eating behavior. The fifth pathway—the incertohypothalamic pathway—forms con-nections from the medial zona incerta to the hypothalamus and the amygdala. It appears to regulate the anticipatory motiva-tional phase of copulatory behavior in rats.
After dopamine was identified as a neurotransmitter in 1959, it was shown that its effects on electrical activity in central synapses and on production of the second messenger cAMP synthesized by adenylyl cyclase could be blocked by antipsychotic drugs such as chlorpromazine, haloperidol, and thiothixene. This evidence led to the conclusion in the early 1960s that these drugs should be considered dopamine-receptor antagonists and was a key factor in the development of dopamine hypothesis of schizophrenia described earlier. The antipsychotic action is nowthought to be produced (at least in part) by their ability to block the effect of dopamine to inhibit the activity of adenylyl cyclase in the mesolimbic system.
B. Dopamine Receptors and Their Effects
At present, five dopamine receptors have been described, con-sisting of two separate families, the D1-like and D2-like receptor groups. The D1 receptor is coded by a gene on chromosome 5, increases cAMP by Gs -coupled activation of adenylyl cyclase, and is located mainly in the putamen, nucleus accumbens, and olfactory tubercle and cortex. The other member of this family, D5, is coded by a gene on chromosome 4, also increases cAMP, and is found in the hippocampus and hypothalamus. The thera-peutic potency of antipsychotic drugs does not correlate with their affinity for binding to the D1 receptor (Figure 29–3, top)
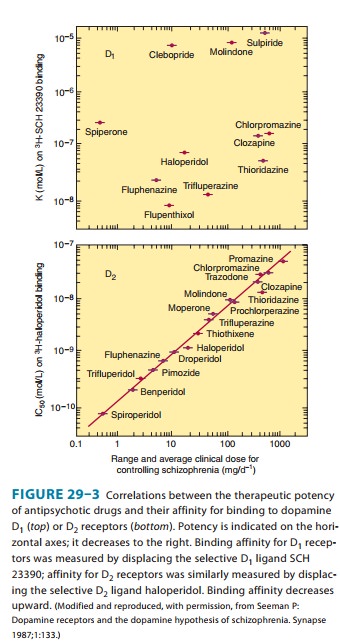
The D2 receptor is coded on chromosome 11, decreases cAMP (by Gi -coupled inhibition of adenylyl cyclase), and inhibits calcium channels but opens potassium channels. It is found both pre- and post-synaptically on neurons in the caudate-putamen, nucleus accumbens, and olfactory tubercle. A second member of this family, the D3 receptor, also coded by a gene on chromosome 11, is thought to also decrease cAMP and is located in the fron-tal cortex, medulla, and midbrain. D4receptors also decrease cAMP and are concentrated in the cortex.
The typical antipsychotic agents block D2 receptors stereoselec-tively for the most part, and their binding affinity is very strongly correlated with clinical antipsychotic and extrapyramidal potency (Figure 29–3, bottom). In vivo imaging studies of D2-receptor occupancy indicate that for antipsychotic efficacy, the typical anti-psychotic drugs must be given in sufficient doses to achieve at least 60% occupancy of striatal D2 receptors. This is not required for the atypical antipsychotic drugs such as clozapine and olanzapine, which are effective at lower occupancy levels of 30–50%, most likely because of their concurrent high occupancy of 5-HT2A receptors. The typical antipsychotic drugs produce EPS when the occupancy of striatal D2 receptors reaches 80% or higher.
Positron emission tomography (PET) studies with aripiprazole show very high occupancy of D2 receptors, but this drug does not cause EPS because it is a partial D2-receptor agonist. Aripiprazole also gains therapeutic efficacy through its 5-HT2A antagonism and possibly 5-HT1A partial agonism.
These findings have been incorporated into the dopamine hypothesis of schizophrenia. However, additional factors complicate interpretation of dopamine receptor data. For example, dopamine receptors exist in both high- and low-affinity forms, and it is not known whether schizophrenia or the antipsychotic drugs alter the proportions of receptors in these two forms.
Ît has not been convincingly demonstrated that antagonism of any dopamine receptor other than the D2 receptor plays a role in the action of antipsychotic drugs. Selective and relatively spe-cific D1, D3-, and D4-receptor antagonists have been tested repeatedly with no evidence of antipsychotic action. Most of the newer atypical antipsychotic agents and some of the traditional ones have a higher affinity for the 5-HT2A receptor than for the D2 receptor (Table 29–1), suggesting an important role for the serotonin 5-HT system in the etiology of schizophrenia and the action of these drugs.
C. Differences among Antipsychotic Drugs
Although all effective antipsychotic drugs block D2 receptors, the degree of this blockade in relation to other actions on receptors varies considerably among drugs. Vast numbers of ligand-receptor binding experiments have been performed in an effort to discover a single receptor action that would best predict antipsychotic efficacy. A summary of the relative receptor-binding affinities of several key agents in such com-parisons illustrates the difficulty in drawing simple conclusions from such experiments:
Chlorpromazine: α1= 5-HT2A> D2> D1
Haloperidol: D2> α1> D4> 5-HT2A> D1> H1
Clozapine: D4= α1> 5-HT2A> D2= D1
Olanzapine: 5-HT2A> H1> D4> D2> α1> D1
Aripiprazole: D2= 5-HT2A> D4> α1= H1>> D1
Quetiapine: H1> α1> M1,3> D2> 5-HT2A
Thus, most of the atypical and some typical antipsychotic agents are at least as potent in inhibiting 5-HT2 receptors as they are in inhibiting D2 receptors. The newest, aripiprazole, appears to be a partial agonist of D2 receptors. Varying degrees of antagonism of α2 adrenoceptors are also seen with risperidone, clozapine, olanzapine, quetiapine, and aripiprazole.
Current research is directed toward discovering atypical antipsychotic compounds that are either more selective for the mesolimbic system (to reduce their effects on the extrapyrami-dal system) or have effects on central neurotransmitter receptors—such as those for acetylcholine and excitatory amino acids—that have been proposed as new targets for anti-psychotic action.
In contrast to the difficult search for receptors responsible for antipsychotic efficacy, the differences in receptor effects of various antipsychotics do explain many of their toxicities (Tables 29–1 and 29–2). In particular, extrapyramidal toxicity appears to be consis-tently associated with high D2 potency.
D. Psychological Effects
Most antipsychotic drugs cause unpleasant subjective effects in nonpsychotic individuals. The mild to severe EPS, including akathisia, sleepiness, restlessness, and autonomic effects are unlike any associated with more familiar sedatives or hypnotics.
Nevertheless, low doses of some of these drugs, particularly que-tiapine, are used to promote sleep onset and maintenance, although there is no approved indication for such usage.
People without psychiatric illness given antipsychotic drugs, even at low doses, experience impaired performance as judged by a number of psychomotor and psychometric tests. Psychotic individuals, however, may actually show improvement in their performance as the psychosis is alleviated. The ability of the atypical antipsychotic drugs to improve some domains of cog-nition in patients with schizophrenia and bipolar disorder is controversial. Some individuals experience marked improve-ment, and for that reason, cognition should be assessed in all patients with schizophrenia and a trial of an atypical agent considered, even if positive symptoms are well controlled by typical agents.
E. Electroencephalographic Effects
Antipsychotic drugs produce shifts in the pattern of electroencepha-lographic (EEG) frequencies, usually slowing them and increasing their synchronization. The slowing (hypersynchrony) is sometimes focal or unilateral, which may lead to erroneous diagnostic interpre-tations. Both the frequency and the amplitude changes induced by psychotropic drugs are readily apparent and can be quantitated by sophisticated electrophysiologic techniques. Some of the neurolep-tic agents lower the seizure threshold and induce EEG patterns typical of seizure disorders; however, with careful dosage titration, most can be used safely in epileptic patients.
F. Endocrine Effects
Older typical antipsychotic drugs, as well as risperidone and pali-peridone, produce elevations of prolactin, see Adverse Effects, below. Newer antipsychotics such as olanzapine, quetiapine, and aripiprazole cause no or minimal increases of prolactin and reduced risks of extrapyramidal system dysfunction and tardive dyskinesia, reflecting their diminished D2antagonism.
G. Cardiovascular Effects
The low-potency phenothiazines frequently cause orthostatic hypotension and tachycardia. Mean arterial pressure, periph-eral resistance, and stroke volume are decreased. These effects are predictable from the autonomic actions of these agents (Table 29–2). Abnormal electrocardiograms have been recorded, especially with thioridazine. Changes include prolongation of QT interval and abnormal configurations of the ST segment and T waves. These changes are readily reversed by withdraw-ing the drug. Thioridazine, however, is not associated with increased risk of torsades more than other typical antipsychot-ics, whereas haloperidol, which does not increase QTc, is. Among the newest atypical antipsychotics, prolongation of the QT or QTc interval has received much attention. Because this was believed to indicate an increased risk of dangerous arrhythmias, approval of sertindole has been delayed and ziprasidone and que-tiapine are accompanied by warnings. There is, however, no evi-dence that this has actually translated into increased incidence of arrhythmias.
H. Animal Screening Tests
Inhibition of conditioned (but not unconditioned) avoidance behavior is one of the most predictive tests of antipsychotic action. Another is the inhibition of amphetamine- or apomorphine-induced stereotyped behavior. Other tests that may predict antipsychotic action are reduction of exploratory behavior without undue sedation, induction of a cataleptic state, inhibition of intracranial self-stimulation of reward areas, and prevention of apomorphine-induced vomiting. Most of these tests are difficult to relate to any model of clinical psychosis.
The psychosis produced by phencyclidine (PCP) has been used as a model for schizophrenia. Because this drug is an antagonist of the NMDA glutamate receptor, attempts have been made to develop antipsychotic drugs that work as NMDA agonists. Sigma receptor and cholecystokinin type b (CCKb) antagonism have also been suggested as potential targets. Thus far, NMDA receptor-based models have pointed to agents that modulate glutamate release as potential antipsychotics. 5-HT2A inverse agonists such as pimavanserin, ritanserin, and M100907 are potent inhibitors of PCP-induced locomotor activity, whereas D2 antagonists are rela-tively weak in comparison. Thus, atypical antipsychotic drugs that act as 5-HT2A antagonists appear much more potent than typical antipsychotic drugs in PCP models.
Related Topics