Chapter: Essential Clinical Immunology: Immunological Aspects of Endocrine Disease
Animal Models of Insulin-Dependent Diabetes Mellitus
ANIMAL MODELS
Five main animal models of IDDM have been described.
The NOD Mouse
This mouse strain is characterized by the onset of IDDM at three or four months of age. Disease incidence and age of onset vary according to gender (with a marked female predominance) and to the sanitary conditions in which the mice are bred (the cleaner the facilities, the higher the disease incidence).
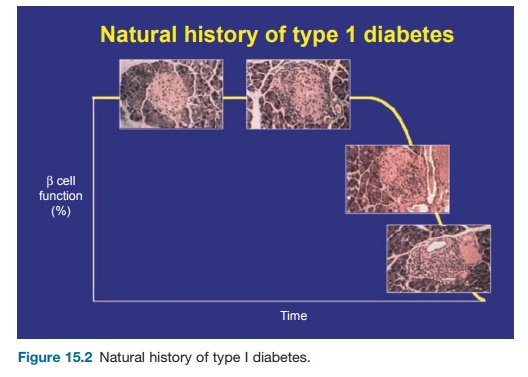
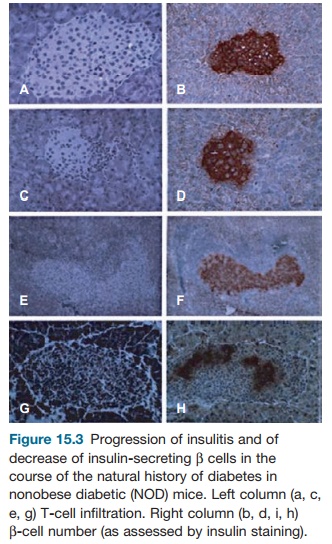
Diabetes onset is preceded for several weeks by insulitis (cellular infiltration of the pancreas, including T cells and mac-rophages), starting at three weeks of age. At six to eight weeks of age, islet-specific autoantibodies and T cells are detected (see Figures 15.2 and 15.3). NOD mice, particu-larly females, do not develop only IDDM. They also often show thyroiditis and siali-tis as well as various extrapancreatic auto-antibodies (essentially antierythrocytic and antinuclear antibodies).
The relevance of the NOD mouse model to human IDDM is a matter of debate. Some minor differences are seen with the human disease: the female predominance is not observed in humans, except in the less common form of IDDM associated with extrapancreatic autoantibodies. Islet-spe-cific antibodies are less prevalent in NOD mice than in human diabetes. In fact, with these two reservations, all other clinical, biological, and immunological characteris-tics of NOD mouse and human IDDM are remarkably similar. The only troublesome point is the unexpected sensitivity of NOD mice to immunointervention. More than 150 therapeutic maneuvers have been shown to prevent diabetes onset when
The Bio-breeding Rat
The bio-breeding (BB) rat is historically the first known spontaneous model of IDDM. It also resembles human IDDM with, how-ever, a major difference, which consists of a major state of lymphocytopenia selectively involving the T-cell subset of Rt6+ cells. Most immunodiabetology laboratories now use the NOD mouse rather than the BB rat because it is easier to handle, it breeds very well, and there are many more genetic and immunological markers avail-able in the mouse than in the rat. However, remarkable studies have been performed in the BB rat, notably the Rossini groups, including the demonstration of IDDM prevention by administration of antilym-phocyte serum and neonatal thymectomy that played a considerable role in drawing attention to the autoimmune origin of the disease.
T-Cell Receptor Transgenic Mice
Several islet-specific CD4+ and CD8+ dia-betogenic T-cell clones have been derived from diabetic NOD mice. When the T-cell receptor (TCR) genes of such clones were used to generate transgenic NOD mice, IDDM developed in an accelerated fash-ion. The most popular mice produced with this strategy were the BDC2.5 mouse (for CD4 T-cell clones) and the 8.1 mouse (for a CD8+ T-cell clone). A large proportion of T cells (but not all) in such mice express the transgenic TCR. This is usually suffi-cient for transgenic mice to develop rapid diabetes. This is not, however, an absolute rule, since in some genetic backgrounds the disease develops slowly or does not even appear. In all cases, the disease is remarkably accelerated after backcross-ing to severe combined immunodeficiency (SCID) or Rag knock out −/− mice that lack both B and T cells, an observation explained by the lack in such backcrossed mice of T cells having undergone endog-enous TCR rearrangements (which prob-ably include regulatory T cells) that nor-mally delay diabetes onset. In fact, T cells from these backcrossed mice are monoclo-nal, all expressing the diabetogenic TCR.
RIP Mice
When transgenes are coupled to the rat insulin promoter (RIP), they are selec-tively expressed in the β cells. This obser-vation has allowed the generation of a wide variety of transgenic models of IDDM. The T antigen of the Simian virus (SV) virus was expressed in β cells of non-autoimmune-prone mice. Depending on individual mice, animals developed insu-linoma or major insulitis (not sufficiently severe, however, to induce IDDM).
Later on, other antigens were used, including influenza virus hemagglutinin (HA) and lymphochoriomeningitis virus (LCMV) glycoprotein. The expression of high amounts of these antigens did not induce diabetes by themselves because the mice were tolerant to the antigens, which, in that context, behaved as bona fide autoantigens. Diabetes could be induced, however, when mice were infected by LCMV (in the case of the RIP-LCMV mice) or when they were crossed to other transgenic mice expressing an HA-specific TCR (in the case of RIP-HA mice). Double transgenic mice were also produced with RIP-LCMV mice, but they did not become diabetic unless they were infected by LCMV.
The Low-Dose Streptozotocin Model
Streptozotocin (STZ) is a selective β-cell toxic agent. Administered at high doses, it induces the quasi-immediate apoptosis of all β cells with subsequent IDDM. When administered at low doses in a repeated fashion, toxic IDDM does not appear. Diabetes is, however, observed after a few days because of autoimmune aggression of the pancreas. Neonatal thymectomy and anti-T-cell antibody treatment prevent low-dose STZ-induced IDDM. This IDDM model has the great advantage of being rapidly induced and obtained in a large variety of mouse strains. Not all strains are equally sensitive to STZ in a geneti-cally controlled fashion. STZ doses must be selected with caution because only a small difference between toxic and immune-trig-gering doses is apparent. The mechanisms leading to STZ-induced diabetes are ill defined but probably involve T-cell trig-gering by β-cell autoantigens released by apoptotic β cells.
Related Topics