Chapter: Modern Pharmacology with Clinical Applications: Antiepileptic Drugs
Sodium Channel Blocking Agents
Sodium Channel Blocking Agents
Drugs sharing this mechanism
include phenytoin (Di-lantin),
carbamazepine (Tegretol),
oxcarbazepine (Tri-leptal),
topiramate (Topamax), valproic acid (Depakene), zonisamide (Zonegran),
and lamotrigine (Lamictal). All of
these agents have the capacity to block sustained high-frequency repetitive
firing (SRF) of action poten-tials. This is accomplished by reducing the
amplitude of sodium-dependent action potentials through an en-hancement of
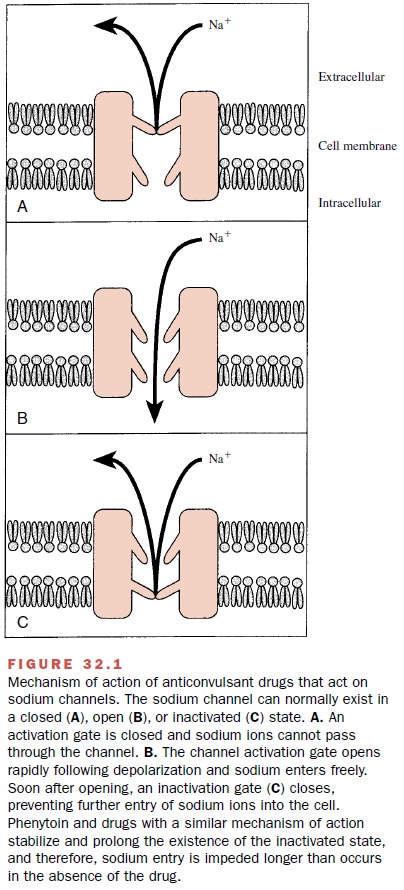
steady-state inactivation. The sodium channel exists in three main
conformations: a resting (R) or activatable state, an open (0) or conducting
state, and an inactive (I) or nonactivatable state. The anticonvul-sant drugs
bind preferentially to the inactive form of the channel. Because it takes time
for the bound drug to dis-sociate from the inactive channel, there is time
depen-dence to the block. Since the fraction of inactive chan-nels is increased
by membrane depolarization as well as by repetitive firing, the binding to the
I state by antiepileptic drugs can produce voltage-, use-, and time-dependent
block of sodium-dependent action potentials. This effect is similar to that of
local anesthetic drugs and is shown in
Figure 32.1.
These agents are discussed
together because their pharmacological properties, clinical indications for the
treatment of epilepsy, and presumed mechanisms of ac-tion are similar. They
differ from each other in several ways, however, and one drug cannot routinely
be sub-stituted for another. They differ primarily in their phar-macokinetic
properties, their adverse reactions, and their interactions with other drugs.
In addition to block-ing sodium channels, some possess other therapeutically
relevant mechanisms of action as well.
Phenytoin
Phenytoin is a valuable agent for the treatment of gen-eralized tonic–clonic seizures and for the treatment of partial seizures with complex symptoms. The establish-ment of phenytoin (at that time known as diphenylhy-dantoin) in 1938 as an effective treatment for epilepsy was more than simply the introduction of another drug for treatment of seizure disorders. Until that time the only drugs that had any beneficial effects in epilepsy were the bromides and barbiturates, both classes of compounds having marked CNS depressant properties.
The prevailing view among neurologists of that era was that epilepsy was the result of excessive electrical activity in the brain and it
therefore seemed perfectly rea-sonable that CNS depressants would be effective
in an-tagonizing the seizures. Consequently, many patients re-ceived high doses
of barbiturates and spent much of their time sedated. Also, since CNS
depression was con-sidered to be the mechanism of action of AEDs, the
pharmaceutical firms were evaluating only compounds with profound CNS
depressant properties as potential antiepileptic agents. It was, therefore,
revolutionary when phenytoin was shown to be as effective as pheno-barbital in
the treatment of epilepsy without any signif-icant CNS depressant activity.
This revolutionized the search for new anticonvulsant drugs as well as
immedi-ately improving the day-to-day functioning of epileptic patients.
An understanding of
absorption, binding, metabo-lism, and excretion is more important for phenytoin
than it is for most drugs. Following oral administration, phenytoin absorption
is slow but usually complete, and it occurs primarily in the duodenum.
Phenytoin is highly bound (about 90%) to plasma proteins, primarily plasma
albumin. Since several other substances can also bind to albumin, phenytoin
administration can displace (and be displaced by) such agents as thyroxine,
tri-iodothyronine, valproic acid, sulfafurazole, and salicylic acid.
Phenytoin is one of very few
drugs that displays zero-order (or saturation) kinetics in its metabolism. At
low blood levels the rate of phenytoin metabolism is proportional to the drug’s
blood 1evels (i.e., first-order kinetics). However, at the higher blood levels
usually required to control seizures, the maximum capacity of drug-metabolizing
enzymes is often exceeded (i.e., the enzyme is saturated), and further
increases in the dose of phenytoin may lead to a disproportionate increase in
the drug’s blood concentration. Since the plasma levels continue to increase in
such a situation, steady-state lev-els are not attained, and toxicity may
ensue. Calculation of half-life (t1/2) values for phenytoin often is
meaning-less, since the apparent half-life varies with the drug blood level.
Acute adverse effects seen
after phenytoin adminis-tration usually result from overdosage. They are
gener-ally characterized by nystagmus, ataxia, vertigo, and diplopia
(cerebellovestibular dysfunction). Higher doses lead to altered levels of
consciousness and cogni-tive changes.
A variety of idiosyncratic
reactions may be seen shortly after therapy has begun. Skin rashes, usually
morbilliform in character, are most common. Exfoliative dermatitis or toxic
epidermal necrolysis (Lyellís syndrome) has been observed but is infrequent.
Other rashes occasionally have been reported, as have a variety of blood
dyscrasias and hepatic necrosis.
The most common side effect
in children receiving long-term therapy is gingival hyperplasia, or over-growth
of the gums (occurs in up to 50% of patients). Although the condition is not
serious, it is a cosmetic problem and can be very embarrassing to the patient.
Hirsutism also is an annoying side effect of phenytoin, particularly in young
females. Thickening of subcuta-neous tissue, coarsening of facial features, and
enlarge-ment of lips and nose (hydantoin facies) are often seen in patients
receiving long-term phenytoin therapy.
Peripheral neuropathy and
chronic cerebellar degener-ation have been reported, but they are rare.
There is evidence that phenytoin
is teratogenic in humans, but the mechanism is not clear. However, it is known
that phenytoin can produce a folate deficiency, and folate deficiency is
associated with teratogenesis.
Only a few well-documented
drug combinations with phenytoin may necessitate dosage adjustment.
Coadministration of the following drugs can result in elevations of plasma
phenytoin levels in most patients: cimetidine, chloramphenicol, disulfiram,
sulthiame, and isoniazid (in slow acetylators). Phenytoin often causes a decline
in plasma carbamazepine levels if these two drugs are given concomitantly.
Ethotoin and mephenytoin are
congeners of pheny-toin that are marketed as AEDs in the United States. They
are not widely used.
Carbamazepine
Carbamazepine has become a
major drug in the treat-ment of seizure disorders. It has high efficacy, is
well tol-erated by most patients, and exhibits fewer long-term side effects
than other drugs.
Oral absorption of
carbamazepine is quite slow and often erratic. Its half-life is reported to
vary from 12 to 60 hours in humans. The development of blood level as-says has
markedly improved the success of therapy with this drug, since serum
concentration is only partially dose related. Carbamazepine is metabolized in
the liver, and there is evidence that its continued administration leads to
hepatic enzyme induction. Carbamazepine-10,11-epoxide is a pharmacologically
active metabolite with significant anticonvulsant effects of its own.
Carbamazepine is an effective
agent for the treat-ment of partial seizures and generalized tonic–clonic
seizures; its use is contraindicated in absence epilepsy. Carbamazepine is also
useful in the treatment of trigeminal neuralgia and is an effective agent for
the treatment of bipolar disorders .
Like most of the agents that
block sodium channels, side effects associated with carbamazepine
administra-tion involve the central nervous system (CNS). Drowsiness is the
most common side effect, followed by nausea, headache, dizziness,
incoordination, vertigo, and diplopia. These effects occur particularly when
the drug is first taken, but tolerance often develops over a few weeks. There
appears to be little risk of cognitive im-pairment with carbamazepine.
Carbamazepine causes a
variety of rashes and other allergic reactions including fever,
hepatosplenomegaly, and lymphadenopathy, but the incidence of serious
hy-persensitivity reactions is rare. Systemic lupus erythe-matosus can occur,
but discontinuation of the drug leads to eventual disappearance of the
symptoms. Idiosyncratic hematological reactions to carbamazepine may occur, but
serious blood dyscrasias are rare. Carbamazepine has been shown to exacerbate
or pre-cipitate seizures in some patients, particularly those ex-hibiting
generalized atypical absences.
While the number of side
effects may be fairly large, most are not serious and can be managed. Severe
ad-verse reactions occur less commonly than with pheny-toin and similar drugs.
The overall incidence of toxicity seems to be fairly low at usual therapeutic
doses.
Most of the drug interactions
with carbamazepine are related to its effects on microsomal drug metabo-lism.
Carbamazepine can induce its own metabolism (autoinduction) after prolonged
administration, de-creasing its clearance rate, half-life, and serum concen-trations.
The possibility of autoinduction requires the clinician to reevaluate the
patient’s blood levels after a month of carbamazepine therapy. The
autoinduction phenomenon is over in about a month.
Carbamazepine also can induce
the enzymes that metabolize other anticonvulsant drugs, including phenytoin,
primidone, phenobarbital, valproic acid, clonazepam, and ethosuximide, and
metabolism of other drugs the patient may be taking. Similarly, other drugs may
induce metabolism of carbamazepine; the end result is the same as for
autoinduction, and the dose of carbamazepine must be readjusted. A common
drug–drug interaction is between carbamazepine and the macrolide antibiotics
erythromycin and trolean-domycin. After a few days of antibiotic therapy, symp-toms
of carbamazepine toxicity develop; this is readily reversible if either the
antibiotic or carbamazepine is discontinued.
Cimetidine, propoxyphene, and
isoniazid also have been reported to inhibit metabolism of carbamazepine. It is
essential to monitor blood levels and adjust the dose if necessary whenever
additional drugs are given to patients taking carbamazepine.
Oxcarbazepine
Oxcarbazepine is chemically
and pharmacologically closely related to carbamazepine, but it has much less
capacity to induce drug-metabolizing enzymes. This property decreases the
problems associated with drug interactions when oxcarbazepine is used in
combination with other drugs. The clinical uses and adverse effect profile of
oxcarbazepine appear to be similar to those of carbamazepine.
Lamotrigine
Lamotrigine has a broad
spectrum of action and is ef-fective in generalized and partial epilepsies. Its
primary mechanism of action appears to be blockage of voltage-dependent sodium
channels, although its effectiveness against absence seizures indicates that
additional mech-anisms may be active. Lamotrigine is almost completelyabsorbed
from the gastrointestinal tract, and peak plasma levels are achieved in about 2
to 5 hours. The plasma half-life after a single dose is about 24 hours. Unlike
most drugs, lamotrigine is metabolized primarily by glucuronidation. Therefore,
it appears likely that lamotrigine will not induce or inhibit cytochrome P450
isozymes, in contrast to most AEDs.
Severe skin rashes appear to
be the major concern with lamotrigine use. The incidence of rash is greater in
children than in adults. Other adverse effects are similar to those of drugs
with the same mechanism of action, such as cerebellovestibular changes leading
to dizziness, diplopia, ataxia, and blurred vision. Disseminated in-travascular
coagulation has been reported.
Topiramate
Topiramate is most useful in
patients with generalized tonic–clonic seizures and those with partial complex
seizures. Topiramate causes a higher incidence of CNS-related side effects
(primarily cognitive slowing and confusion) than other AEDs. It does not appear
to cause a significant incidence of rashes or other hyper-sensitivity
reactions; however, a significantly higher in-cidence of kidney stones has been
observed in persons receiving topiramate than in a similar untreated
popu-lation.
Zonisamide
Zonisamide has only recently
been approved for use in the United States, although it has been available in
Japan for several years. It is effective in partial complex and generalized tonic–clonic
seizures and also appears to be beneficial in certain myoclonic seizures. It
has a long half-life (about 60 hours) and requires about 2 weeks to achieve
steady-state levels. It causes cere-bellovestibular side effects similar to
those of most other AEDs sharing its mechanism of action. In addi-tion, it
appears to cause an increased incidence of kid-ney stones.
Valproic Acid (Sodium Valproate)
Although it is marketed as
both valproic acid (Depakene) and as
sodium valproate (Depakote), it is
the valproate ion that is absorbed from the gastroin-testinal tract and is the
active form.
As with several other AEDs,
it is difficult to ascribe a single mechanism of action to valproic acid. This
com-pound has broad anticonvulsant activity, both in exper-imental studies and
in the therapeutic management of human epilepsy. Valproic acid has been shown
to block voltage-dependent sodium channels at therapeutically relevant
concentrations. In several experimental stud-ies, valproate caused an increase
in brain GABA; the mechanism was unclear.There is evidence that valproate may
also inhibit T-calcium channels and that this may be important in its mechanism
of action in patients with absence epilepsy.
Valproic acid is well
absorbed from the gastroin-testinal tract and is highly bound (~90%) to plasma
pro-tein, and most of the compound is therefore retained within the vascular
compartment. Valproate rapidly en-ters the brain from the circulation; the
subsequent de-cline in brain concentration parallels that in plasma,
in-dicating equilibration between brain and capillary blood. A large number of
metabolites have been identi-fied, but it is not known whether they play a role
in the anticonvulsant effect of the parent drug. Valproic acid inhibits the
metabolism of several drugs, including phe-nobarbital, primidone,
carbamazepine, and phenytoin, leading to an increased blood level of these
compounds. At high doses, valproic acid can inhibit its own metabo-lism. It can
also displace phenytoin from binding sites on plasma proteins, with a resultant
increase in un-bound phenytoin and increased phenytoin toxicity. In this
instance, the dosage of phenytoin should be ad-justed as required. These
examples reinforce the need to determine serum anticonvulsant levels in
epileptic patients when polytherapy is employed.
Valproic acid has become a
major AED against sev-eral seizure types. It is highly effective against
absence seizures and myoclonic seizures. In addition, valproic acid can be used
either alone or in combination with other drugs for the treatment of
generalized tonic– clonic epilepsy and for partial seizures with complex
symptoms.
The most serious adverse
effect associated with val-proic acid is fatal hepatic failure. Fatal
hepatotoxicity is most likely to occur in children under age 2 years,
espe-cially in those with severe seizures who are given multi-ple
anticonvulsant drug therapy. The hepatotoxicity is not dose related and is
considered an idiosyncratic re-action; it can occur in individuals in other age
groups, and therefore, valproic acid should not be administered to patients
with hepatic disease or significant hepatic dysfunction or to those who are
hypersensitive to it. Valproic acid administration has been linked to an
in-creased incidence of neural tube defects in the fetus of mothers who
received valproate during the first trimester of pregnancy. Patients taking
valproate may develop clotting abnormalities.
Valproic acid causes hair
loss in about 5% of pa-tients, but this effect is reversible. Transient
gastroin-testinal effects are common, and some mild behavioral effects have
been reported. Metabolic effects, including hyperglycemia, hyperglycinuria, and
hyperammonemia, have been reported. An increase in body weight also has been
noted. Valproic acid is not a CNS depressant, but its administration may lead
to increased depression if it is used in combination with phenobarbital,
primi-done, benzodiazepines, or other CNS depressant agents.
Related Topics