Chapter: Paediatrics: Bones and joints
Paediatrics: Osteogenesis imperfecta
Osteogenesis imperfecta
An inherited condition affecting
collagen matura-tion and organization. The incidence is around 1/20 000.
Osteogenesis im-perfecta (OI) is due to a mutation in type I collagen gene that
predisposes to fracture formation. Following a fracture, initial bone healing
is normal, but there is no subsequent remodelling and the bone heals with
deformity. 10% are clinically asymptomatic.
Clinical features
ŌĆó
Bones:
ŌĆó
low
birth weight/length for gestational age;
ŌĆó
short
stature;
ŌĆó
50%
scoliosis.
ŌĆó
Joints: ligamentous laxity resulting in
hyperextensible joints.
ŌĆó
Specific
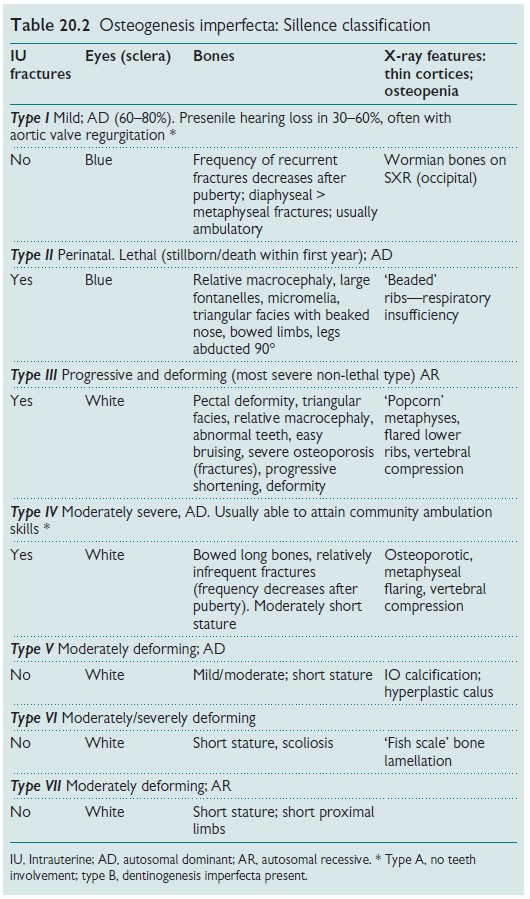
signs and X-ray features (see Table 20.2).
Investigations and diagnosis
ŌĆó
Prenatal
US scan may detect severe forms in foetus.
ŌĆó
Molecular
genetic testing (pre- or postnatal).
ŌĆó
Biochemistry: normal/increased alkaline
phosphatase (ALP).
ŌĆó
Skin biopsy: assess collagen in cultured
fibroblasts.
ŌĆó
Bone biopsy: histologyŌĆöincreased Haversian
canal + osteocyte lacunae diameters,
increased cell numbers.
Treatment
No curative treatment. Aim to
prevent and manage frac-tures with long-term rehabilitation.
Prevention
Strategies to decrease fracture
frequency include:
ŌĆó
oral
calcium supplements;
ŌĆó
bisphosphonates;
ŌĆó
synthetic
calcitonin.
Surgical interventions
Intramedullary rods (fixed
length/telescoping) to prevent bowing of long bones, especially for fractures
in children >2yrs old. Corrective surgery for scoliosis deformities >50┬░.
Prognosis
In severe OI a good predictor of future walking is being able to sit by 10mths. May develop cardiopulmonary or neurological complications. Usually develop progressive shortening and deformity caused by multiple fractures, e.g. ŌĆśsabreŌĆÖ tibia, ŌĆśaccordionŌĆÖ femora.
Related Topics