Chapter: Essentials of Psychiatry: Sedative-Hypnotic Agents
Sedative-Hypnotic Agents: Prescription Medications
Prescription
Medications
Prescription
medications for patients suffering from insomnia gen-erally should be used only
on a short-term basis. Benzodiazepines replaced barbiturates as the mainstay of
treatment for much of the past 30 years. Benzodiazepines have a decreased abuse
potential, fewer interactions with other drugs, a broader therapeutic index
compared with the barbiturates, and pose a much lower risk in overdose. The
adverse effect profile of benzodiazepines includes the risk for abuse and
physiological dependence that develops with daily use, as well as problems with
daytime sedation, motor incoordination and cognitive impairment. More recently
the imi-dazopyridine, zolpidem and the pyrazolopyrimidine, zaleplon, have
become increasingly popular as daytime sedation and the risk for abuse and
physiological dependence appear to be some-what less frequent with these two
agents.
Benzodiazepines
Benzodiazepines
are clearly effective for transient and situ-ational insomnias. A recent
meta-analysis demonstrated that benzodiazepine treated patients reported
improvement in sleep onset, number of awakenings at night, the total amount of
sleep obtained, and the quality of sleep compared with placebo-treated patients
(Nowell et al., 1997). Although only
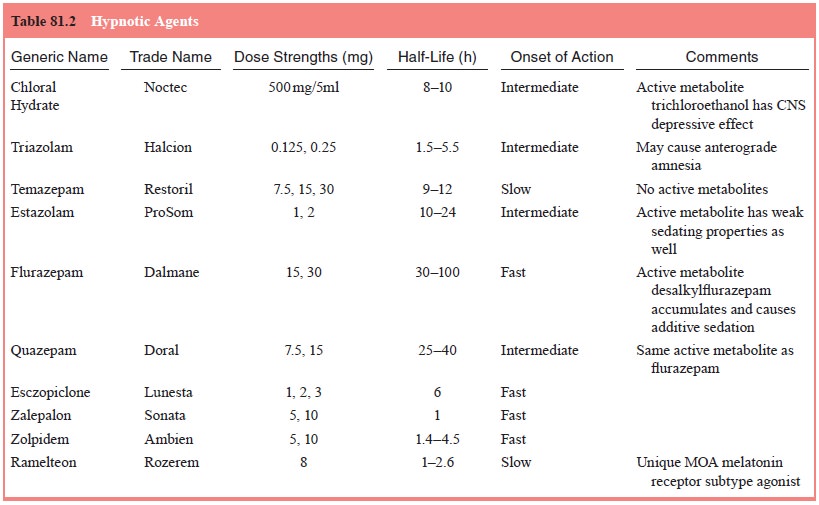
five benzodiazepines are marketed as hypnotic agents (Table 81.2), any
benzodiazepine could be used to induce sleep provided an appropriate dose is
chosen. Benzodiazepines improve sleep by inducing drowsiness, relaxing muscles
and decreasing mental agitation. As doses, and consequently brain
concentrations, are increased, drowsi-ness and relaxation shift into decreased
wakefulness and then sleep. Benzodiazepines increase total sleep time, increase
non-rapid-eye-movement (non-REM) sleep, decrease sleep latency,
decrease
stage 1 sleep and increase stage 2 sleep. Effects on stage 3 sleep vary based
on the individual drug used, but stage 4 sleep is generally reduced.
Benzodiazepines are central nervous system depressants. Their likely mechanism of action relates to their ability to aug-ment the opening of neuronal gamma-aminobutyric acid receptor-related chloride channels. By modulating the effects of gamma-aminobutyric acid, benzodiazepines increase the frequency of chloride channel openings. This effect is in contrast to the effects of barbiturates or alcohol, which seem to increase the duration of chloride channel opening. While this distinction may appear minor, it accounts for the greater safety of benzodiazepines in overdose (i.e., there is less likelihood of respiratory depression or coma). When alcohol is mixed with a benzodiazepine overdose, the synergistic effects of respiratory depression can have poten-tially fatal consequences.
There is
little convincing evidence that the five marketed benzodiazepine hypnotics
differ in terms of efficacy or safety when they are administered appropriately
(i.e., amounts, dos-ing intervals and duration of use). The current
benzodiazepines marketed as hypnotic agents are similar in their effects on
sleep architecture and differ only in onset of action and duration of action.
The individual agent chosen should be based on the type of sleep difficulty
being experienced, the age of the patient and comorbid diagnoses. Triazolam has
a rapid onset and a short duration of action, making it a better choice for
patients with sleep initiation difficulties, while flurazepam has a somewhat
longer onset of action and a much longer duration of action, making it a better
choice for patients with middle or terminal insomnia.
Shorter
half-life benzodiazepine hypnotics cause less daytime sedation and residual
cognitive affects the day follow-ing administration. Rebound insomnia often
occurs with these agents, however, especially after use for several consecutive
nights. Rebound insomnia can usually be avoided by taper-ing the dose or using
lower dosages during treatment. There is some evidence to suggest that the likelihood
of rebound in-somnia is greater in patients who experience greater hypnotic
efficacy.
Besides rebound insomnia, some patients receiving shorter half-life benzodiazepines have problems with middle or terminal insomnia. Plasma levels are quite low several hours after tak-ing these medications and, as a result, if there is a problem with sleep maintenance these agents are not as likely to be as helpful as hypnotics with longer half-lives. In addition, shorter half-life benzodiazepines can sometimes cause an increase in daytime anxiety, particularly in the morning. For at least some patients, this may reflect the development of physiologic dependence and a withdrawal phenomenon.
Shorter
half-life hypnotics, particularly triazolam, ap-pear to cause anterograde
amnesia more commonly than the longer half-life agents. Numerous reports in the
lay press re-garding a greatly increased risk of hallucinations, confusion and
anterograde amnesia with triazolam use does not appear to be supported by an
examination of all available data. All benzodi-azepines can cause anterograde
amnesia, however, particularly at higher dosages. Anterograde amnesia commonly
occurs follow-ing a benzodiazepine overdose. The prevalence of anterograde
amnesia with appropriate dosing of sedative–hypnotic agents and risk factors
for its development in patients are still open questions at this time.
Longer
half-life benzodiazepines may be more appropri-ate for patients who suffer from
middle or terminal insomnia since these agents retain significant sedative
properties for many hours after initial administration. Some patients may have
problems with daytime sedation or decreased reaction time the day following
administration. Sedative agents such as flurazepam and quazepam that have
active metabolites with long half-lives are particularly prone to causing such
problems. The active metabolite often builds up over time causing an in-crease
in daytime drowsiness, decreased reaction time and in coordination with
repeated administration, particularly in the elderly. Patients with significant
daytime anxiety may benefit from the longer acting benzodiazepines as the
residual amount of medication left in the morning may still have anxiolytic
benefits.
The
elderly use benzodiazepine hypnotic agents to a much greater degree than the
general adult population. While this is likely due to the greater prevalence of
sleep disorders in the elderly, the use of hypnotic agents in this population
poses particular challenges. The elderly as a group have a decrement in
hepatically metabolized benzodiazepines (e.g., estazolam, flurazepam, quazepam,
triazolam), which is often more pro-nounced among men. Incoordination, sedation
and confusion can occur when such agents build up over time. Ray and
col-leagues found a much higher rate of falls and hip fractures in the elderly
population that received long-acting benzodiazepine hypnotics. The risk of hip
fracture increased in direct propor-tion to the daily dose of the long-acting
benzodiazepine. Short-acting benzodiazepines were not found to have a similar
prob-lem in their two large case–control studies (Ray et al., 1987, 1989). A generally safe clinical strategy when
managing insom-nia problems in the elderly is to halve the starting dose and
ti-trate up slowly as tolerated following the longstanding advice “start low,
go slow”. Avoiding long-acting benzodiazepines ap-pears prudent as well.
Drug
interactions with benzodiazepines are quite com-mon. When mixed with other
sedating compounds the effects are additive. Narcotic medications, alcohol and
antihistamines are examples where the additive sedation can lead to confusion
quite easily. In addition, acute alcohol ingestion slows hepatic metabolism and
causes transiently higher concentrations of oxidatively metabolized
benzodiazepines leading to further se-dation. Antacids that slow gastric
emptying may decrease the rate of absorption of a hypnotic agent from its
primary site of origin, the small intestine. This could alter both onset and
peak concentration effects.
Benzodiazepines
all undergo some form of hepatic metab-olism, although some agents are much
more extensively metabo-lized than others. Medications that inhibit hepatic
microsomal oxidative metabolism may cause clinically meaningful drug
in-teractions when combined with benzodiazepines. All currently available
triazolobenzodiazepines (e.g., alprazolam, estazolam, midazolam, triazolam) and
diazepam are metabolized by hepatic microsomes and are complete or partial
substrates of the CYP 3A4 isoform. Medications that are potent inhibitors of
this system will cause higher peak concentrations of the
triazolobenzodiazepine, particularly triazolam, which is highly hepatically
metabolized. Nefazodone, ketaconazole, cimetidine and macrolide antibiotics are
examples of clinically relevant CYP 3A4 inhibitors which may cause a reduction
in the clearance of these triazolobenzodi-azepines and an increase in their
blood and brain concentrations. This increase is greatest with higher hepatic
clearance drugs such as triazolam or midazolam and somewhat less for drugs such
as alprazolam or estazolam.
All
benzodiazepine hypnotic agents are FDA Pregnancy Category X, meaning their use
should be avoided during preg-nancy, especially during the first trimester due
to an increased risk of congenital malformations. A variety of congenital
malfor-mations, including cleft palate, delayed ossification of a number of
bony structures and an increased occurrence of rudimentary ribs have been
reported.
Benzodiazepines
must be used in patients with obstructive sleep apnea only with extreme
caution. Benzodiazepines may cause respiratory depression and can render
patients less likely to mount an appropriate respiratory response to hypoxia.
Hypnotic agents such as zolpidem, zaleplon, or trazodone are less likely to
cause problems for sleep apnea patients and may be preferable alternatives.
Aggressive evaluation and treatment of sleep apnea [e.g., (continuous positive
airway pressure) CPAP] is extremely important to ensure adequate restorative
sleep.
Chloral Hydrate
Chloral
hydrate was among the earliest sleeping pills. Its sedating qualities become
evident within 30 minutes of adminis-tration, as it is rapidly absorbed from
the gastrointestinal tract. At dosages between 0.5 and 1.5 g, it is an
effective hypnotic agent. The elimination half-life of its active metabolite,
trichloroetha-nol, is 6 to 8 hours, rendering it unlikely to cause significant
next-day sedation or functional impairment. Despite the potential ben-efits of
a rapid onset of action and relatively short elimination half-life, chloral
hydrate is rarely used as a hypnotic agent today. It has a narrow therapeutic
index (toxic dose–therapeutic dose), causes gastric irritation, nausea and
vomiting easily, and may cause gastric necrosis at high doses. Overdose can be
fatal due to respiratory depression.
Zolpidem
Zolpidem
is also a nonbenzodiazepine hypnotic. A member of the imidazopyridine class, it
is available in 5 and 10 mg dosages. Its mechanism of action shares much in
common with benzo-diazepines as it is active at central benzodiazepine
(sometimes called ω)
receptors. The ω receptor
is a subunit of the GABAAreceptor. Benzodiazepines are thought to
bind nonselectively and activate all ω receptor
subtypes; by contrast, zolpidem ap-pears to bind preferentially to ω1
receptors. Although this selec-tive binding is not absolute, it may explain the
relative absence of myorelaxant, anxiolytic and anticonvulsant effects of
zolpidem at hypnotic dose, as well as the preservation of stages 3 and 4 sleep.
Polysomnographic experience with zolpidem indicates that zolpi-dem induces a
sleep pattern very similar to that of physiological sleep, and generally
produces little effect on sleep architecture following abrupt discontinuation
(Darcourt et al., 1999).
Zolpidem
is rapidly absorbed through the gastrointestinal tract and has a rapid onset of
action. Peak concentration occurs from 30 minutes to 2 hours following
administration. It is me-tabolized in the liver to several inactive metabolites
and has an elimination half-life of approximately 2.5 hours. The elimination
half-life is prolonged in the elderly and in patients with impaired hepatic or
renal function. Zolpidem overdose can cause respira-tory depression or coma,
especially when combined with other CNS depressants. The benzodiazepine
antagonist flumazenil can reverse the effects of an overdose of zolpidem,
reflecting its ben-zodiazepine-like mechanism of action.
Although
zolpidem is classified as a schedule IV drug by the FDA, it appears to cause
tolerance and withdrawal syn-dromes somewhat less frequently than
benzodiazepine hypnotics do. Withdrawal symptoms occur more frequently at doses
higher than listed in the Physician’s
Desk Reference (PDR), which rec-ommends a maximum dose of 10 mg. Cross
tolerance with al-cohol and benzodiazepines are found at higher doses, and the
incidence of adverse effects is much higher. Anecdotal reports of
hallucinations and confusion at standard hypnotic dosages have been reported
(Toner et al., 1999, Ansseau et al., 1992), but fur-ther research is
needed to determine the prevalence of such oc-currences across a spectrum of
dosages and age groups.
Zaleplon
Zaleplon
is a nonbenzodiazepine hypnotic of the pyrazolopyrimi-dine class. Like
zolpidem, zaleplon binds preferentially to central benzodiazepine (or ω) receptors. Zaleplon binds to
the ω1, ω2 and ω3 subunits, while zolpidem bind
only to the ω1 subunit. Zaleplon is rapidly absorbed from the
gastrointestinal tract, reaching peak serum concentrations within 1 hour.
Absolute bioavailability is only 30% as it undergoes extensive hepatic
first-pass metabolism. Its elimination half-life is approximately 1 hour. Since
cimeti-dine inhibits aldehyde oxidase and CYP 3A4, and may increase zaleplon
plasma concentrations by 85%, the initial starting dose of zaleplon should be
halved in patients also taking cimetidine. Other potent inhibitors of the CYP
3A4 system such as ketocona-zole and erythromycin also may increase zaleplon
levels.
Because
of the quick onset of action of zaleplon and its short elimination half-life,
it can be used for patients who have a difficult time initiating sleep, but are
able to remain asleep throughout the remainder of the night. In patients who
have dif-ficulty both in initiating and in maintaining sleep, zaleplon may
permit the initiation of sleep, but be less effective in maintaining sleep due
to its short elimination half-life. On the other hand, this same short
half-life can be an advantage, as it can permit middle of the night dosing for
those patients who awaken then. The short half-life permits such a dosing
schedule without causing signifi-cant next-day sedation (Walsh et al., 2000).
Zaleplon,
like zolpidem, is classified as a schedule IV drug by the FDA. Higher than
standard dosages have been associated with an abuse potential similar to that
of triazolam. With dosages of 20 mg or less, abuse and dependence appears to be
signifi-cantly less than that of benzodiazepine hypnotic agents (Rush et al.,
1999). Rebound insomnia has been reported, but it appears to occur less frequently than in the short acting benzodiazepines
such as triazolam.
Other Prescription Hypnotics
Barbiturates
are commonly prescribed outside the USA for in-somnia. Butabarbital, phenobarbital
and secobarbital are the most commonly prescribed barbiturates. The
barbiturates are poten-tially lethal in overdose due to the risk of respiratory
depression, and they commonly cause induction of hepatic oxidative metabo-lism.
Their use has generally been replaced by other agents with better side-effect
profiles such as the benzodiazepines, the ima-dazopyridines and the
pyrazolopyrimidines.
Hydroxyzine
hydrochloride and hydroxyzine pamoate are sedating H1 receptor
antagonists that are used occasionally as hypnotic agents. Sedation occurs with
these agents due to inhi-bition of histamine N-methyltransferase and blockage of central histaminergic
receptors. Next-day sedation is a common problem with antihistamines as they
have a relatively long elimination half-life. These agents are highly
anticholinergic and may cause hypotension and are not usually recommended as
first-line agents for the treatment of insomnia.
Trazodone,
a triazolopyridine compound marketed as an antidepressant, is frequently used
as a hypnotic. Hypnotic dos-ages vary markedly, from 25 to 300 mg, depending on
individual susceptibility to its sedating effects. Trazodone increases slow
wave sleep and total sleep time and does not appear to affect REM sleep, unlike
other antidepressants (Yamadera et al.,
1998). Its elimination half-life of between 6 and 9 hours renders trazo-done
likely to cause daytime drowsiness. Using the lowest effec-tive dosage can
minimize this effect, as taking the medication in late evening rather than at
bedtime. Trazodone is commonly used to counter the insomnia associated with
SSRI use. It is rarely used as a sole antidepressant because of its strong
sedat-ing qualities and the need to take the medication more than once per day
because of its half-life. Tolerance to its sedating effects develops only
rarely with long-term use, making it an excellent option for those with chronic
insomnia. Priapism is an exceed-ingly rare side effect, occurring in less than
one in 40 000 cases. Since anticholinergic side effects and postural
hypotension are not common, trazodone has some advantages over the tricyclic
antidepressants.
Related Topics