Chapter: Pathology: Immunopathology
Primary Immune Deficiency Syndromes
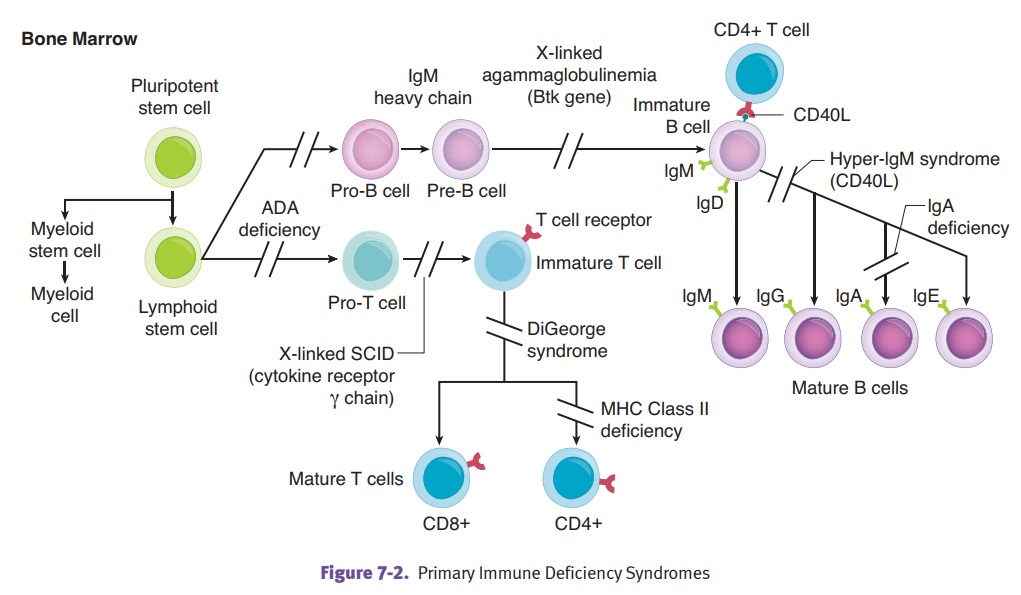
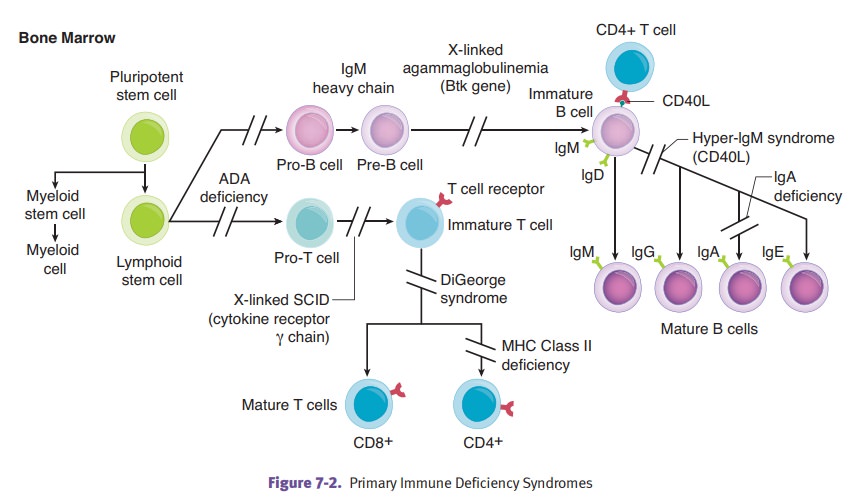
PRIMARY IMMUNE DEFICIENCY SYNDROMES
X-linked
agammaglobulinemia of Bruton is an immunodeficiency characterized
bya developmental failure to produce mature B cells and plasma cells, resulting
in agammablobulinemia. The condition occurs because of loss of function
mutations of B-cell Bruton tyrosine kinase (BTK). Clinically, the disease
affects male infants who have recurrent infections beginning at 6 months of
life due to the loss of passive maternal immunity. Common infections include
pharyngitis, otitis media, bronchi-tis, and pneumonia; common infecting
organisms include H. influenza, S.
pneumo-coccus, and S. aureus.
Common
variable immunodeficiency is a group of disorders
characterized by defectsin B-cell maturation that can lead to defective IgA or
IgG production. Clinically, both sexes are affected with onset in childhood of
recurrent bacterial infections and with increased susceptibility to Giardia lamblia. Complications include
increased frequency of developing autoimmune disease, non-Hodgkin lymphoma, and
gastric cancer.
DiGeorge
syndrome is an embryologic failure to develop the 3rd and 4th
pharyngealpouches, resulting in the absence of the parathyroid glands and
thymus. Clinical findings can include neonatal hypocalcemia and tetany, T-cell
deficiency, and recur-rent infections with viral and fungal organisms.
Severe
combined immunodeficiency (SCID) is a combined deficiency of
cell-mediatedand humoral immunity that is often caused by a progenitor-cell
defect. The modes of inheritance are variable and can include X-linked
(mutation of the common [gamma] chain of the interleukin receptors IL-2, IL -4,
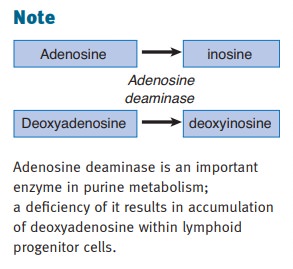
IL-7, IL-9, IL-15, and IL-21) and autosomal recessive (deficiency of adenosine
deaminase). Clinical features include recurrent infections with bacteria,
fungi, viruses, and protozoa; susceptibil-ity to Candida, cytomegalovirus (CMV), and Pneumocystis jirovecii infections, and adverse reactions to live
virus immunizations. SCID is treated with hematopoietic stem cell transplantation
since the prognosis without treatment is death of most infants within a year.
Wiskott-Aldrich
syndrome is an X-linked recessive disease with mutation in the
genefor Wiskott-Aldrich syndrome protein (WASP). The disease has a clinical
triad of recurrent infections, severe thrombocytopenia, and eczema (chronic
spongiform dermatitis). Treatment is hematopoietic stem cell transplantation.
Complications include increased risk of non-Hodgkin lymphoma and death due to
infection or hemorrhage.
Complement
system disorders can involve a variety of factors,
with deficiencies ofdifferent factors producing different clinical patterns.
In
both the classical and alternate pathways, C3 deficiency causes both recurrent
bacterial infections and immune complex disease, while C5, C6, C7, and C8
deficien-cies cause recurrent meningococcal and gonococcal infections.
·
In the classical pathway only, C1q,
C1r, C1s, C2, and C4 deficiencies cause marked increases in immune complex
diseases, including infections with pyogenic bacteria.
·
In the alternate pathway, Factor B
and properdin deficiencies cause increased neisserial infections. Deficiencies
in complement regulatory proteins can cause C1-INH deficiency (hereditary
angioedema), which is characterized clinically by edema at mucosal surfaces
with low C2 and C4 levels.
MHC class
II deficiency can be caused by defects in positive selection of
thymocytes.Few CD4+ lymphocytes develop and as a result, patients suffer from
severe immuno-deficiency. Mutations in genes (i.e., CIITA) that encode proteins that regulate MHC class II gene
expression are the cause. CD8+ T cells are unaffected.
Hyper IgM
syndrome is characterized by normal B and T lymphocyte numbers
andnormal to elevated IgM levels but significantly decreased IgA, IgG and IgE
levels. Mutations in the gene for CD40 ligand result in the most common form of
X-linked hyper IgM syndrome.
Selective
IgA deficiency has unknown genetic etiology. Many affected individu-als
appear healthy while others have significant illness. Sinopulmonary infections,
diarrhea and adverse reactions to transfusions can occur. Levels of IgA are
unde-tectable whereas levels of other isotypes are normal. There is an
association with autoimmune disease.
Related Topics