Chapter: Essentials of Psychiatry: Psychiatric Pathophysiology: Mood Disorders
Monoamine Alterations
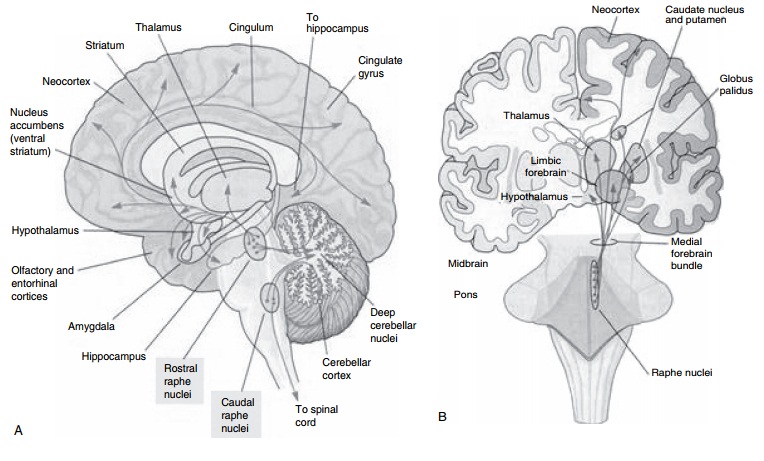
Monoamine Alterations
Serotonergic System
Serotonin has been implicated in the
pathophysiology of depression and bipolar disorder (for reviews, see Mann,
1999; Oquendo and Mann, 2000; Shiah and Yatham, 2000; Mann et al., 2001; Nemeroff et al.,
1997; Meltzer and Lowy, 1987; Coppen, 1969; Mace and Taylor, 2000; Kandel, 2000; Charney et al., 1981) (Table 11.1 and Figure 11.1). This hypothesis
proposed that the vulnerability to either depression or mania was related to
low serotonergic activity, attrib-utable to either less serotonin release or
fewer serotonin receptors or impaired serotonin receptor-mediated signal
transduction. Prange et al., (1974) formulated a permissive
hypothesis of serotonin function in
bipolar disorder. They suggested that a deficit in central seroton-ergic neurotransmission
permits the expression of bipolar disorder, and that both the manic and
depressive phases of bipolar disorder are characterized by low central
serotonergic neurotransmission. Over the last 30 years, a variety of studies of
the serotonergic sys-tem have reinforced its role in major depression and
identified ad-ditional associations with suicidal behavior, impulsivity,
aggression, eating disorders, obsessive–compulsive disorder, anxiety disorders,
personality disorders, seasonal changes in mood and behavior, and alcohol abuse
and dependence. The serotonergic system also plays a role in the regulation of
a variety of basic biological functions in-cluding sleep, appetite, circadian
rhythm and cognitive function.
Medications that target the serotonin transporter
site and se-lectively inhibit reuptake of serotonin (e.g., fluoxetine,
sertraline, paroxetine, fluvoxamine, citalopram) have all been shown to be
effective antidepressants (Nemeroff et al.,
1997; Sampson, 2001;

Mace and Taylor, 2000). Some antidepressant drugs
specifically act at one of the many serotonin receptor subtypes. For example,
suggested antidepressive/anxiolytic medications buspirone and gepirone are 5-HT1A
receptor agonists, and fewer 5-HT1A receptors are implicated in the
pathophysiological mechanism of depression and anxiety (Yocca, 1990; Apter and
Allen, 1999).
Considered together, studies of serotonin function
in major depression suggest both hypofunction and likely compensatory changes
that would increase serotonergic activity (Brown et al., 1994; Leonard, 1994; Dubovsky and Buzan, 1999). Findings
such as 1) lower serotonin and 5-HIAA levels in postmortem brain stem and lower
CSF 5-HIAA; 2) relapse of depression with diet acute depletion of tryptophan;
3) fewer serotonin transporter sites in prefrontal cortex; 4) fewer
postsynaptic 5-HT1A receptors; and 5) the antidepressant properties
of medications that enhance serotonergic transmission suggest that
underactivity of the serotonin system is part of the pathogenesis of
depression. Conversely, more 5-HT2A receptor binding in the frontal
cortex of depressed individuals who committed suicide, fewer brainstem 5-HT1A
autoreceptors and fewer serotonin transporters in the raphe nuclei would tend
to increase serotonergic transmission in major depression. There is evidence
for the contribution of serotonin in mania and in the mechanism of action of
mood stabilizers (Shiah and Yatham, 2000); however, the data on the role of the
serotonergic system in mania are fewer and not consistent. Alterations in
functioning of other neurotransmitters in mania such as norepinephrine, dopamine,
acetylcholine and GABA, and their interaction with serotonin may also
contribute. Future studies of serotonergic activity in mood disorders will need
further to differentiate primary pathogenesis from compensatory changes.
Noradrenergic System
There are multiple lines of evidence that the
noradreneric system is disordered in depression (Berman et al., 1996; Charney, 1998; He-ninger et al., 1996; Leonard, 1997; Owens, 1997; Kandel, 2000; Pot-ter et al., 1993; Schatzberg and
Schildkraut, 1995; Nemeroff et al.,
1997; Ressler and Nemeroff, 1999) (Table 11.2 and Figure 11.2).

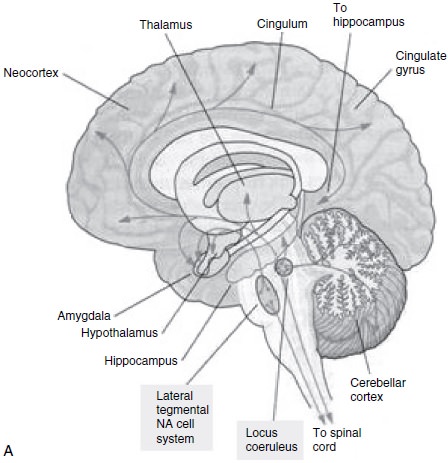
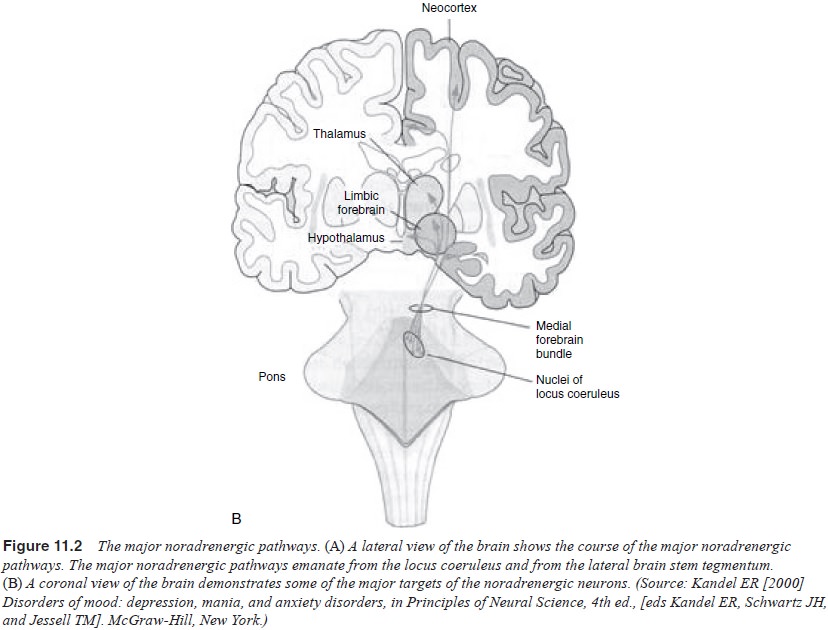
A large body of metabolite data are consistent with
the hy-pothesis that there are abnormalities in the noradrenergic system in
depression. The conflicting findings, however, are not consist-ent with simple
increased or decreased noradrenergic activity.
Dopaminergic System
Some studies have found that CSF levels of the
dopamine metabolite homovanillic acid (HVA) are lower in patients with major
depression than in controls and that lower CSF HVA levels are found in more
severely depressed patients (see Kapur and Mann, 1992; Brown et al.,
1994, for a summary) (Table 11.3). However, other studies failed to replicate these findings or found
higher CSF HVA in patients with depression (Jimerson, 1987; Vestergaard et al., 1978).

Gamma-aminobutyric Acid
Gamma-aminobutyric acid (GABA) is the major
inhibitory neu-rotransmitter in almost all areas of the CNS and regulates many
CNS functions (Nemeroff et al.,
1997). A decrease in GABAergic activity may play a role in depression by
regulating receptor re-sponses to catecholamines (Enna et al., 1986; Nemeroff et al.,
1997). Pathophysiological contributions of GABA and thera-peutic effects of
GABAergic medications in mood disorders may be mediated via effects on other
neurotransmitter systems (Dubovsky and Buzan, 1999; Nemeroff et al., 1997).
Other Neurotransmitters
Cholinergic neurons containing acetylcholine
project diffusely throughout the cortex (Thase, 2000). The involvement of
cholinergic system in the pathogenesis of depression is supported by the
following findings: cholinergic input reduces REM latency (decreased REM
la-tency is seen in depression); some antidepressants have anticholiner-gic
properties; lecithin, an acetylcholine precursor, reduces mania in some cases
and can induce depression; and cholinergic rebound fol-lowing abrupt withdrawal
of anticholinergic medications can cause a relapse of depression (Dubovsky and
Buzan, 1999; Dilsaver and Coffman, 1989; Janowsky and Risch, 1984; Keshavan,
1985).
There are emerging data that drugs that antagonize
NMDA receptors have antidepressant effects (Przegalinski et al., 1997; Papp and Moryl, 1994; Trullas and Skolnick, 1990).
It is important to note that all neurotransmitters
and receptors interact with and influence each other (Brown et al., 1994; Leonard, 1994; Dubovsky
and Buzan, 1999). Most cerebral functions are the result of the converging
action of many different neurotransmitters. It is not likely that the
pathophysiology of mood disorders is due to a single neurotransmitter (Brown et al., 1994). More probably, mood
disorders are disorders of the overall interaction of multiple transmitter
systems. Alternatively, different components of depres-sion may be related to
different neurotransmitter dysfunction.
The binding of a neurotransmitter to a postsynaptic
receptor triggers a cascade of chemical processes that include the second
messenger systems (Thase, 2000; Dubovsky and Buzan, 1999; Thase and Howland,
1995). The bidirectional actions of second messengers allow unitary changes in
second messenger function to produce diverse changes in transmitter synthesis
and release, and in receptor activity, leading to complex neurotransmitter and
receptor effects. There is evidence that mood-stabilizing drugs (e.g., lithium)
act upon G proteins or other second messengers (Jesberger and Richardson, 1985;
Kofman and Belmaker, 1993; Wang et al.,
2001; Chen et al., 2001).
Related Topics