Chapter: Medical Immunology: Tumor Immunology
Immunogenicity of Tumors
IMMUNOGENICITY OF TUMORS
The immunogenicity of tumors has been a major topic of debate for several decades. In a seminal study on the immune response induced by spontaneous tumors, it was reported that none of 27 spontaneous murine tumors tested exhibited any detectable immunogenicity. However, the results of this study contrasted with the well-documented observation that ro-dent tumors induced by chemical carcinogens can elicit an immune response that causes re-jection of tumor grafts in syngeneic animals. It is now accepted that even spontaneously oc-curring tumors induced in laboratory animals can be immunogenic when tested under appropriate experimental conditions.
Similarly, it was initially assumed that human cancers are less immunogenic than murine tumors; however, additional experimental work has shown that many human can-cers are antigenic, even when the host immune response does not appear to prevent tumor progression. Some of the discrepancies between animal studies and human studies may re-sult from experimental limitations. For example, it has been argued that only through trans-plantation of tumors might one be able to clearly demonstrate immunogenicity of certain human cancers. Obviously, such experiments cannot be performed.
A. Rejection antigens
Rejection antigens were originally defined as those responsible for the induction of an ef-fective immunity against murine tumors induced by the chemical carcinogen methyl-cholanthrene (MCA). Similar observations were subsequently made with other chemical carcinogens, and, interestingly, rejection antigens appear to differ among different car-cinogens and also among different animals given the same carcinogen; in other words, they seem to be unique to each individual tumor.
These observations suggested that a mutational mechanism, not necessarily directed to the same genetic target, might have been responsible for the generation of antigenic entities easily recognized by the host immune response. At the same time, it was shown that tumor rejection was mostly mediated through the cellular arm of the immune system (T cells). In vitro studies utilizing cancer-specific T cells also revealed that the recogni-tion of antigenic entities by lymphocytes did not necessarily correlate with tumor rejec-tion in vivo.
These experimental results led to the important conclusion that antigenicity and im-munogenicity are not synonyms; in other words, antigenicity of a tumor per se may not be sufficient to ensure rejection of the same tumor.
B. Tumor-Associated Antigens
The realization from animal studies that antigens expressed by chemically induced tumors were unique gave the impression that any potential approach to cancer immunotherapy in humans might have been hopeless. However, later studies also indicated that virally in-duced as well as spontaneous neoplasms, including human cancers, did express shared tu-mor antigens, raising the possibility that immunotherapeutic approaches to human cancer could be devised and tested. However, it is important to point out that unique rejection anti-gens were easily identified by virtue of their ability to induce tumor rejection, suggesting that their individuality correlated with immunogenicity. On the other hand, shared tumor antigens, commonly known as tumor-associated antigens, were identified by different means, since they could not generally induce a measurable immune response directed against the tumor.
Given the practical impossibility of utilizing unique antigens for devising realistic immunotherapeutic approaches for cancer, it is important to fully understand the nature of tumor-associated shared antigens and the biology of any immune response directed against them in order to devise immunotherapeutic strategies for cancer that have a realistic chance of success in a clinical setting.
The mutant ras oncogene–encoded proteins and the mutant p53 suppressor gene–en-coded proteins represent important examples of shared tumor-associated antigens that have undergone mutational changes. In both cases, the mutational changes provide the cell with a growth advantage that eventually results in neoplastic growth; however, there is very lit-tle evidence that epitopes derived from the mutated regions of each protein are strongly im-munogenic. In other words, these tumor-associated antigens do not represent tumor-rejec-tion antigens.
These observations also suggest that environmental carcinogens may be responsible for the mutational changes that occur in spontaneous tumors and that these changes are not very different in nature and mechanism of induction from the mutations that are induced in experimental and murine tumors by well-studied chemical carcinogens like MCA. There-fore, rejection antigens are probably the result not of qualitative differences among muta-tions induced in chemically induced tumors versus spontaneous ones; rather, the large num-ber of mutations found in chemically induced tumors obtained following the exposure of experimental animals to a large dose of carcinogen results in strong immunogenicity.
Additional examples of mutational changes possibly induced by environmental con-ditions are genomic rearrangements resulting in fusion proteins whose expression strongly correlates with the malignant phenotype. For instance, the epidermal growth factor recep-tor (EGF-R) gene undergoes an internal deletion in 40% of malignant glioblastomas, and the resulting altered gene product triggers growth of these tumor cells (unstoppable by cur-rent treatments). Since the mutated protein can be recognized by specific monoclonal anti-bodies, new treatments based on this observation may be tested in the near future for this uniformly lethal condition. At the present time it is not clear whether any tumor-associated antigens resulting from mutational changes like the ones discussed above will provide an effective target for immune-based treatments of those cancers that express them.
Normal cellular proteins overexpressed in certain cancers represent a second group of tumor-associated antigens. Several of these proteins have been identified and character-ized in great detail in human melanoma, which represents possibly the best-studied tumor from an immunological perspective because of the extensive clinical and biological evi-dence that the host immune response can have a measurable impact on the natural course of this disease.
Examples of melanoma-associated, overexpressed tumor antigens are MART-1, GP100, and tyrosinase. These gene products are also lineage-specific, meaning that their expression appears to be limited to the melanocyte lineage, and their overexpression cor-relates with neoplastic development.
The MAGE family of melanoma antigens represents a separate set of melanoma-as-sociated antigens that result from overexpression of normal cellular genes. These proteins are not lineage-specific, being normally expressed only in testes, but they are tumor-spe-cific, because they are often overexpressed not only in melanoma, but also in several other epithelial cancers.
Lineage-specific and MAGE-type tumor-associated antigens have become the object of considerable interest, since the results of several recent clinical studies suggest that well-defined epitopes derived from these antigens when injected in conjunction with incomplete Freund adjuvant into select patients with melanoma could induce measurable biological and sometimes even clinical responses.
Additional studies have led to the identification of lineage-specific antigens in non-melanoma cancer types. The primary example is the carcinoembryonic antigen (CEA), which tends to be overexpressed in most adenocarcinomas of the gastrointestinal tract. An-other important example is the prostate-specific antigen (PSA), a protease expressed by prostate gland cells that becomes elevated in hypertrophic or cancerous prostate.
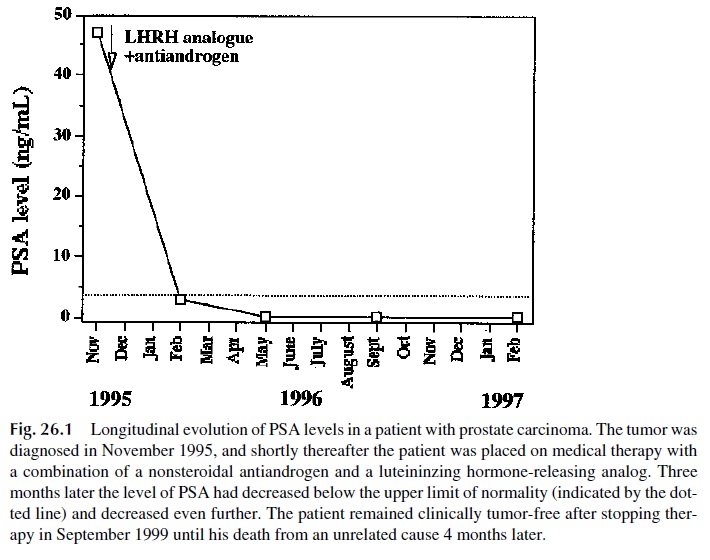
A third example is the alpha-fetoprotein (AFP), which becomes elevated in patients with cancer of the liver or testes. These tumor-associated antigens are often overexpressed and may pro-vide useful markers for monitoring the course of disease, since they may be measured in serum (Fig. 26.1).
Overexpression of the human epidermal growth factor receptor 2 (EGFR-2), also known as HER-2/neu, is observed in some breast and ovarian cancers, and its presence cor-relates with a more aggressive disease. It is worth noting that a monoclonal antibody against this molecule is currently approved for treating breast cancers overexpressing HER-2. The idiotype of the surface immunoglobulin in B-cell malignancies also constitutes a lin-eage-specific tumor-associated antigen, which is also unique and therefore clonal in nature. Clinical studies utilizing monoclonal antibodies to target B-cell idiotypes are currently be-ing tested and show some promise.
Related Topics