Chapter: Modern Pharmacology with Clinical Applications: The Rational Basis for Cancer Chemotherapy
Cancer Chemotherapy: Drug Resistance
DRUG RESISTANCE
Many patients undergoing
chemotherapy fail to re-spond to treatment from the outset; their cancers are
re- sistant to the available agents. Other patients respond initially, only to
relapse.
Cancers can be regarded as
populations of cells un-dergoing spontaneous mutations. The population be-comes
increasingly heterogeneous as the tumor grows and increasing numbers of
mutations occur. Tumors of the same type and size will vary in their
responsiveness to therapy because of the chance occurrences of drug-resistant
mutations during tumor growth.
Assuming the same initial
drug sensitivity, smaller tumors are generally more curable than larger tumors
because of the increased probability of drug-resistant mutations in the larger
tumors. Therefore, therapy ear-lier in
the course of tumor growth should increase the chance for cure. Combination
chemotherapy is often more effective than treatment with single drugs.
Tumors that are resistant to drugs from the outset will always have a largely
drug-resistant population and will be re-fractory to treatment.
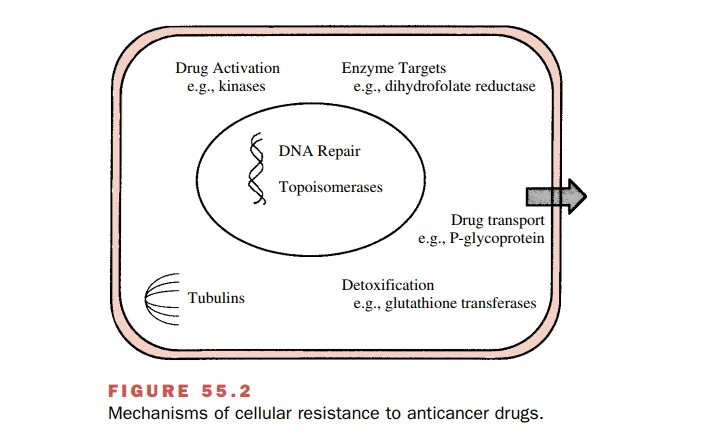
Many kinds of biochemical
resistance to anticancer drugs have been described. The biochemical and genetic
mechanisms of resistance to methotrexate
are now known in some detail. Three major resistance pathways have been
described: (1) decreased drug transport into cells; (2) an alteration in the
structure of the target en-zyme dihydrofolate reductase (DHFR), resulting in
re-duced drug affinity; and (3) an increase in DHFR con-tent of tumor cells.
The increase in DHFR content occurs through a process of gene amplification, that is, a reduplication or increase in the
number of copies per cell of the gene coding for DHFR. Amplification of
var-ious genes may be a relatively frequent event in tumor cell populations and
an important genetic mechanism for generating resistance to drugs.
Tumor cells may become
generally resistant to a va-riety of cytotoxic drugs on the basis of decreased
uptake or retention of the drugs. This form of resistance is termed pleiotropic, or multidrug, resistance, and it is the major form of resistance to
anthracyclines, vinca alka-loids, etoposide, paclitaxel, and dactinomycin. The
gene that confers multidrug resistance (termed mdr I) en-codes a high-molecular-weight membrane protein called P-glycoprotein, which acts as a drug
efflux pump in many tumors and normal tissues.
Possible biochemical
mechanisms of resistance to alkylating
agents include changes in cell DNA repair ca-pability, increases in cell
thiol content (which in turn can serve as alternative and benign targets of
alkylation), decreases in cell permeability, and increased activity of
glutathione transferases. Increased metallothionein content has been associated
with tumor cell resistance to cisplatin.
Drugs that require metabolic
activation for antitu-mor activity, such as the antimetabolites 5-fluorouracil and 6-mercaptopurine, may be
ineffective if a tumor is deficient in the required activating enzymes. Alter-
natively, a drug may be metabolically inactivated by re-sistant tumors, which
is the case with cytarabine (pyrim-idine nucleoside deaminase) and bleomycin
(bleomycin hydrolase). Leukemias have been shown to develop re-sistance to L-asparaginase because of a
drug-related in-duction of the enzyme asparagine synthetase.
Major mechanisms of cellular
resistance to anti-cancer drugs are depicted in Fig. 55.2.
Related Topics