Chapter: Clinical Anesthesiology: Clinical Pharmacology: Analgesic Agents
Analgesic Agents: Opioids
Analgesic Agents
Regardless of how expertly surgical and
anesthetic procedures are performed, appropriate prescrip-tion of analgesic
drugs, especially opioids and cyclooxygenase (COX) inhibitors, can make the
difference between a satisfied and an unsatisfied postoperative patient.
Studies have shown that out-comes can be improved when analgesia is provided in
a “multimodal” format (typically emphasizing COX inhibitors and local
anesthetic techniques while minimizing opioid use) as one part of a
well-defined and well-organized plan for postoperative care .
OPIOIDS
Mechanisms of Action
Opioids bind to specific receptors
located through-out the central nervous system and other tissues. Four major
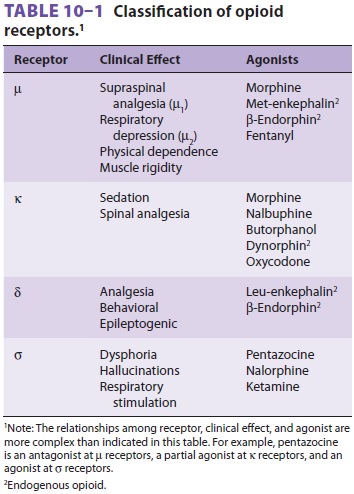
opioid receptor types have been identi-fied ( Table 10–1): mu (µ, with subtypes µ1 and µ2), kappa (κ), delta (δ), and sigma (σ). All opioid recep-tors couple to G
proteins; binding of an agonist to an opioid receptor causes membrane
hyperpolariza-tion. Acute opioid effects are mediated by inhibition of adenylyl
cyclase (reductions in intracellular cyclic
adenosine monophosphate concentrations)
and activation of phospholipase C. Opioids inhibit volt-age-gated calcium
channels and activate inwardly rectifying potassium channels. Opioid effects
vary based on the duration of exposure, and opioid toler-ance leads to changes
in opioid responses.
Although opioids provide some degree of
seda-tion and (in many species) can produce general anesthesia when given in
large doses, they are prin-cipally used to provide analgesia. The properties of
specific opioids depend on which receptor is bound (and in the case of spinal
and epidural administra-tion of opioids, the location in the neuraxis where the
receptor is located) and the binding affinity of the drug. Agonist–antagonists
(eg, nalbuphine, nalorphine, butorphanol, and pentazocine) have less efficacy
than so-called full agonists (eg, fentanyl) and under some circumstances will
antagonize the actions of full agonists.
The opioid drugs mimic endogenous
com-pounds. Endorphins, enkephalins, and dynorphins are endogenous peptides
that bind to opioid recep-tors. These three families of opioid peptides differ
in their amino acid sequences, anatomic distributions, and receptor affinities.
Opioid receptor activation inhibits the
pre-synaptic release and postsynaptic response to excitatory neurotransmitters
(eg, acetylcholine, substance P) from nociceptive neurons. The cel-lular
mechanism for this action was described at the beginning. Transmission of pain
impulses can be selectively modified
at the level of the dorsal horn of the spinal cord with intrathe-cal or
epidural administration of opioids. Opioid receptors also respond to
systemically adminis-tered opioids. Modulation through a descending inhibitory
pathway from the periaqueductal gray matter to the dorsal horn of the spinal
cord may also play a role in opioid analgesia. Although opi-oids exert their
greatest effect within the central nervous system, opiate receptors have also
been identified on somatic and sympathetic peripheral nerves. Certain opioid
side effects (eg, depression of gastrointestinal motility) are the result of
opi-oid binding to receptors in peripheral tissues (eg, the wall of the
gastrointestinal tract), and there are now selective antagonists for opioid
actions out-side the central nervous system (alvimopan and oral naltrexone).
The distribution of opioid recep-tors on axons of primary sensory nerves and
the clinical importance of these receptors (if present) remains speculative,
despite the persisting prac-tice of compounding of opioids in local anesthetic
solutions applied to peripheral nerves.
Structure–Activity Relationships
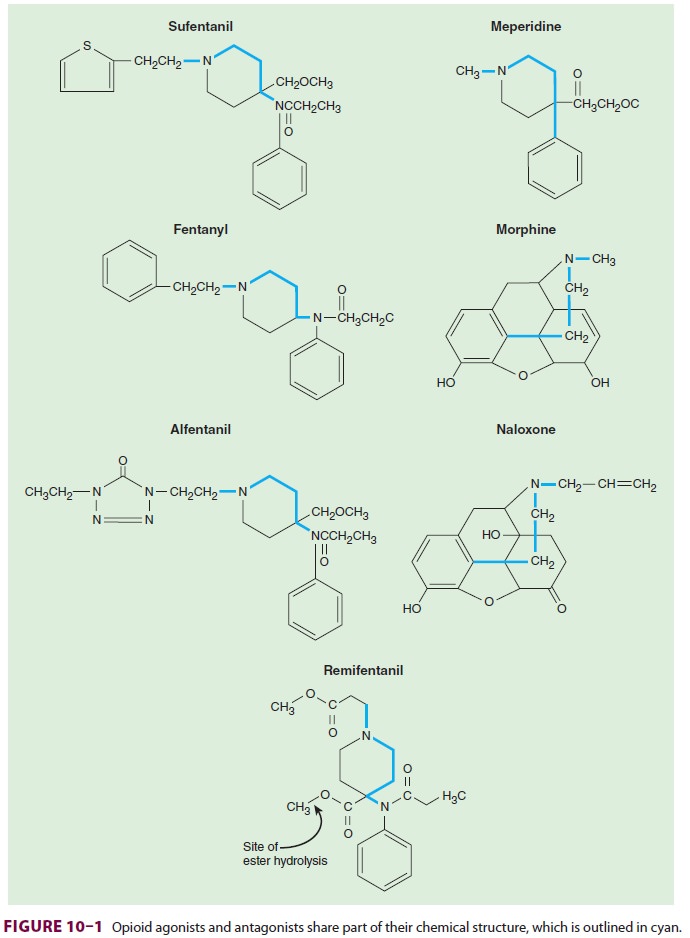
Opioid receptor binding is a property
shared by a chemically diverse group of compounds. Nonethe-less, there are
common structural characteristics, which are shown in Figure 10–1. As is true for most
classes of drugs, small molecular changes can convert an agonist into an
antagonist. The levorotatory iso-mers are generally more potent than the
dextrorota-tory opioid isomers.
Pharmacokinetics
A. Absorption
Rapid and complete absorption follows
the intra-muscular injection of hydromorphone, morphine, or meperidine, with
peak plasma levels usually reached after 20–60 min. Oral transmucosal fentanyl
citrate absorption (fentanyl “lollipop”) provides rapid onset of analgesia and
sedation in patients who are not good candidates for conventional oral,
intravenous, or intramuscular dosing of opioids. The low molecular weight and
high lipid solu-bility of fentanyl also favor transdermal absorption (the
transdermal fentanyl “patch”). The amount of fentanyl absorbed per unit of time
depends on the surface area of skin covered by the patch and also on local skin
conditions (eg, blood flow). The time required to establish a reservoir of drug
in the upper dermis delays by several hours the achievement of effective blood
concentrations. Serum concentra-tions of fentanyl reach a plateau within 14–24
h of application (with peak levels occurring after a lon-ger delay in elderly
than in younger patients) and remain constant for up to 72 h. Continued
absorp-tion from the dermal reservoir accounts for persist-ing measurable serum
levels many hours after patch removal. Fentanyl patches are most often used for
outpatient management of chronic pain and are par-ticularly appropriate for
patients who require con-tinuous opioid dosing but cannot take the much less
expensive, but equally efficacious, oral agents such as methadone.
A wide variety of opioids are effective
by oral administration, including oxycodone, hydrocodone (most often in
combination with acetaminophen), codeine, tramadol, morphine, hydromorphone,
and methadone. These agents are much used for outpa-tient pain management.
Fentanyl is often administered in small doses (10–25 mcg) with local anesthetics for spinal anes-thesia, and adds to the analgesia when included with local anesthetics in epidural infusions. Morphine in doses between 0.1 and 0.5 mg and hydromorphone in doses between 0.05 and 0.2 mg provide 12–18 hours of analgesia after intrathecal administration. Morphine and hydromorphone are commonly included in local anesthetic solutions infused for postoperative epidural analgesia. Extended-release epidural morphine (DepoDur) is administered as a single epidural dose (5–15 mg), the effects of which persist for 48 h.
B. Distribution
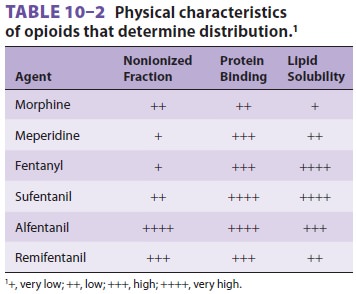
Table 10–2 summarizes
the physical characteris-tics that determine distribution and tissue binding of
opioid analgesics. After intravenous administra-tion, the distribution
half-lives of all of the opioids are fairly rapid (5–20 min). The low fat
solubility of morphine slows passage across the blood–brain bar-rier, however,
so that its onset of action is slow and its duration of action is prolonged.
This contrasts with the increased lipid solubility of fentanyl and sufentanil,
which are associated with a faster onset and shorter duration of action when administeredin small doses.
Interestingly, alfentanil has a morerapid onset of action and shorter duration
of action than fentanyl following a bolus injection, even though it is less
lipid soluble than fentanyl. The high nonionized fraction of alfentanil at
physiological pH and its small volume of distribution ( Vd) increase the amount of drug (as a
percentage of the adminis-tered dose) available for binding in the brain.
Signifi cant amounts of lipid-soluble
opioids can be retained by the lungs (first-pass uptake); as systemic
concentrations fall they will return to the bloodstream. The amount of
pulmonary uptake is reduced by prior accumulation of other drugs, increased by
a history of tobacco use, and decreased by concurrent inhalation anesthetic
administration.
Unbinding of opioid receptors and
redistribu-tion (of drug from effect sites) terminate the clini-cal effects of
all opioids. After smaller doses of the lipid-soluble drugs (eg, fentanyl or
sufentanil), redistribution alone is the driver for reducing blood
concentrations, whereas after larger doses biotrans-formation becomes an important
driver in reducing plasma levels below those that have clinical effects. Thus,
the time required for fentanyl or sufentanil concentrations to decrease by half
is context sensi-tive; in other
words, the half-time depends on thetotal dose of drug and duration of exposure
.
C. Biotransformation
With the exception of remifentanil, all
opioids depend primarily on the liver for biotransformation and are metabolized
by the cytochrome P (CYP) system, conjugated in the liver, or both. Because of
the high hepatic extraction ratio of opioids, their clearance depends on liver
blood flow. The small Vdof alfentanil contributes to a short
eliminationhalf-life (1.5 h). Morphine and hydromorphone undergo conjugation
with glucuronic acid to form, in the former case, morphine 3-glucuronide and
morphine 6-glucuronide, and in the latter case, hydromorphone 3-glucuronide.
Meperidine is N-demethylated to
normeperidine, an active metab-olite associated with seizure activity,
particularly after very large meperidine doses. The end products of fentanyl,
sufentanil, and alfentanil are inactive. Norfentanyl, the metabolite of
fentanyl, can be mea-sured in urine long after the native compound is no longer
detectable in blood to determine chronic fen-tanyl ingestion. This has its greatest
importance in diagnosing fentanyl abuse.
Codeine is a prodrug that becomes active
after it is metabolized by CYP to morphine. Trama-dol similarly must be
metabolized by CYP to O-desmethyltramadol
to be active. Oxycodone ismetabolized by CYP to series of active compounds that
are less potent than the parent one.
The ester structure of remifentanil
makes it sus-ceptible to hydrolysis (in a manner similar to esmo-lol) by
nonspecific esterases in red blood cells and tissue (see Figure 10–1), yielding
a terminal elimi-nation half-life of less than 10 min. Remifentanil
biotransformation is rapid and the duration of a
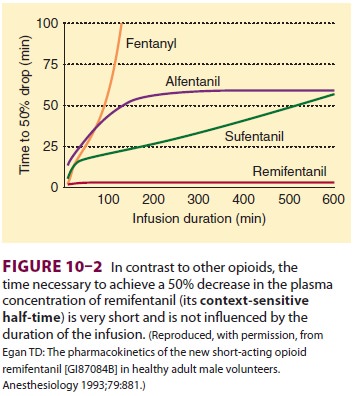
remifentanil infusion has little effect
on wake-up time (Figure 10–2). The context-sensitive half-time of
remifentanil remains approximately 3 min regardless of the dose or duration of
infusion. In its lack of accumulation remifentanil differs from other currently
available opioids. Hepatic dysfunc-tion requires no adjustment in remifentanil
dos-ing. Finally, patients with pseudocholinesterase deficiency have a normal
response to remifentanil (as also appears true for esmolol).
D. Excretion
The end products of morphine and
meperidine bio-transformation are eliminated by the kidneys, with less than 10%
undergoing biliary excretion. Because 5–10% of morphine is excreted unchanged
in the urine, kidney failure prolongs morphine duration of action. The
accumulation of morphine metab-olites (morphine 3-glucuronide and
morphine6-glucuronide) in patients with kidney failure has been associated with
prolonged narcosis and venti-latory depression. In fact, morphine 6-glucuronide
is a more potent and longer-lasting opioid agonist than morphine. As previously
noted, normeperi-dine at increased concentrations may produce sei-zures; these
are not reversed by naloxone. Renal dysfunction increases the likelihood of
toxic effectsfrom normeperidine accumulation. However, both morphine and
meperidine have been used safely and successfully in patients with kidney
failure. Metabolites of sufentanil are excreted in urine and bile. The main
metabolite of remifentanil is elimi-nated in urine, is several thousand times
less potent than its parent compound, and thus is unlikely to produce any
clinical opioid effects.
Effects on Organ Systems
A. Cardiovascular
In general, opioids have few direct
effects on the heart. Meperidine tends to increase heart rate (it is
structur-ally similar to atropine and was originally synthe-sized as an
atropine replacement), whereas larger doses of morphine, fentanyl, sufentanil,
remifentanil, and alfentanil are associated with a vagus nerve– mediated
bradycardia. With the exception of meperi-dine (and only then at very large
doses), the opioids do not depress cardiac contractility provided they are administered
alone (which is almost never the circum-stance in surgical anesthetic
settings). Nonetheless, arterial blood pressure often falls as a result of
bra-dycardia, venodilation, and decreased sympathetic reflexes, sometimes
requiring vasopressor support. These effects are more pronounced when opioids
are administered in combination with benzodiazepines, in which case drugs such
as sufentanil and fentanyl can be associated with reduced cardiac output. Bolus
doses of meperidine, hydromorphone, and morphine evoke histamine release in
some individuals that can lead to profound drops in systemic vascular
resistance and arterial blood pressure. The potential hazards of histamine
release can be minimized in susceptible patients by infusing opioids slowly or
by pretreatment with H1 and H2 antagonists, or both. The end effects of
histamine release can be reversed by infusion of intra-venous fluid and
vasopressors.
Intraoperative hypertension during
large-dose opioid anesthesia or nitrous oxide–opioid anesthesia is common. Such
hypertension is oftenattributed to inadequate anesthetic depth, thus it is
conventionally treated by the addition of other anes-thetic agents
(benzodiazepines, propofol, or potent inhaled agents). If depth of anesthesia
is adequate and hypertension persists, vasodilators or other antihypertensives
may be used. The inherent cardiacstability provided by opioids is greatly
diminished in actual practice when other anesthetic drugs, includ-ing nitrous
oxide, benzodiazepines, propofol, or volatile agents, are typically added. The
end result of polypharmacy can include myocardial depression.
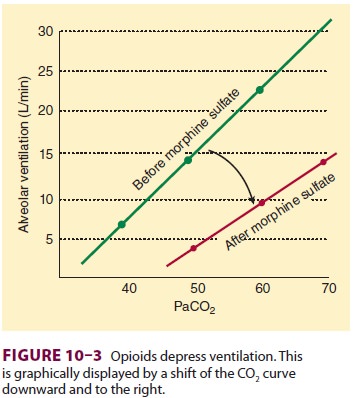
B. Respiratory
Opioids depress ventilation,
particularly respiratory rate. Thus, monitoring of respiratory rate provides a
convenient, simple way of detecting early respiratory depression in patients
receiving opioid analgesia. Opioids increase the partial pressure of carbon
diox-ide (Paco2) and blunt the response to a CO2 chal-lenge, resulting in a shift of the CO2 response curve downward and to the right (Figure
10–3). These effects result from opioid binding to neurons in the respiratory
centers of the brainstem. The apneic threshold—the greatest Paco2 at which a
patient remains apneic—rises, and hypoxic drive is decreased. Morphine and
meperidine can cause histamine-induced bronchospasm in susceptible patients.
Rapid administration of larger doses of opioids (particularly fentanyl,
sufentanil, remifentanil, and alfentanil) can induce chest wall rigidity severe
enough to prevent ade-quate bag-and-mask ventilation. This centrally
mediated muscle contraction is
effectively treated with neuromuscular blocking agents. This problem is rarely
seen now that large-dose opioid anesthesia is less often used in cardiovascular
anesthesia prac-tice. Opioids can effectively blunt the bronchocon-strictive
response to airway stimulation such as occurs during tracheal intubation.
C. Cerebral
The effects of opioids on cerebral
perfusion and intracranial pressure must be separated from any effects of
opioids on Paco2. In general, opioids reduce cerebral
oxygen consumption, cerebral blood flow, cerebral blood volume, and
intracranial pressure, but to a much lesser extent than barbiturates,
pro-pofol, or benzodiazepines. These effects will occur during maintenance of
normocarbia by artificial ventilation; however, there are some reports of mild—
but transient and almost certainly unimportant— increases in cerebral artery
blood flow velocity and intracranial pressure following opioid boluses in
patients with brain tumors or head trauma. If com-bined with hypotension, the
resulting fall in cerebral perfusion pressure could be deleterious to patients with abnormal intracranial
pressure–volume rela-tionships. Nevertheless, the important clinical message is
that any trivial opioid-induced increase in intracranial pressure would likely
be much less important than the much more likely large increases in
intracranial pressure associated with intubation that might be observed in an
inadequately anesthe-tized patient (from whom opioids were withheld). Opioids
usually have almost no effects on the elec-troencephalogram (EEG), although
large doses are associated with slow δ-wave activity. There are curi-ous
sporadic case reports that large doses of fentanyl may rarely cause seizure
activity; however, some of these apparent seizures have been retrospectively
diagnosed as severe opioid-induced muscle rigidity. EEG activation and seizures
have been associated with the meperidine metabolite normeperidine, as previously
noted.
Stimulation of the medullary
chemoreceptor trigger zone is responsible for opioid-induced nausea and
vomiting. Curiously, nausea and vomiting are more common following smaller
(analgesic) than very large (anesthetic) doses of opioids. Prolonged oral
dosing of opioids or infusion of large doses of remi-fentanil during general
anesthesia can produce the phenomenon of opioid-induced tolerance. Repeated
dosing of opioids will reliably produce tolerance, a phenomenon in which larger
doses are required to produce the same response. This is not the same as
physical dependence or addiction, which may also be associated with repeated
opioid administration.Prolonged dosing of opioids can also produce
“opioid-induced hyperalgesia,” in whichpatients become more sensitive to
painful stimuli. Infusion of large doses of (in particular) remifent-anil
during general anesthesia can produce acute tolerance, in which much larger
than usual doses of opioids will be required for postoperative analgesia.
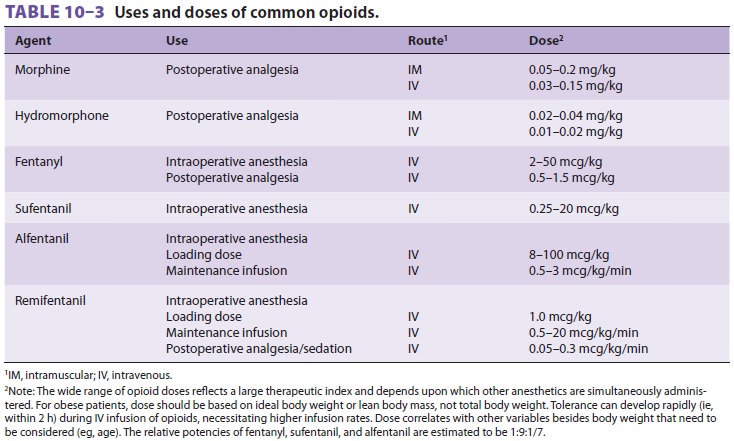
Relatively large doses of opioids are required to ren-der patients unconscious
(Table 10–3).
Regardless of the dose, however, opioids will not reliably pro-duce amnesia.
Parenteral opioids have been the mainstay of pain control for more than a
century. The relatively recent use of opioids in epidural and intrathecal
spaces has revolutionized acute and chronic pain management.
Unique among the commonly used opioids,
meperidine has minor local anesthetic qualities, par-ticularly when
administered into the subarachnoid space. Meperidine’s clinical use as a local
anesthetic has been limited by its relatively low potency and propensity to
cause typical opioid side effects (nau-sea, sedation, and pruritus) at the
doses required to induce local anesthesia. Intravenous meperidine (10–25 mg) is
more effective than morphine or fentanyl for decreasing shivering in the
postanes-thetic care unit and meperidine appears to be the best agent for this
indication.
D. Gastrointestinal
Opioids slow gastrointestinal motility by binding to opioid receptors in the gut and reducing peristalsis. Biliary colic may result from opioid-induced con-traction of the sphincter of Oddi. Biliary spasm, which can mimic a common bile duct stone on cholangiography, is reversed with the opioid antagonist naloxone or glucagon. Patients receiv-ing long-term opioid therapy (eg, for cancer pain) usually become tolerant to many of the side effects but rarely to constipation. This is the basis for the recent development of the peripheral opioid antagonists methylnaltrexone and alvimopan, and for their salutary effects in promoting motility in patients with opioid bowel syndrome, those receiv-ing chronic opioid treatment of cancer pain, and those receiving intravenous opioids after abdomi-nal surgery.
E. Endocrine
The neuroendocrine stress response to
surgi-cal stimulation is measured in terms of thesecretion of specific
hormones, including catechol-amines, antidiuretic hormone, and cortisol. Large
doses of opioids (typically fentanyl or sufentanil) block the release of these
hormones in response to surgery more completely than volatile anesthetics.
Although much discussed, the actual clinical out-come benefit produced by
attenuating the stress response, even in high-risk cardiac patients, remains
speculative (and possibly nonexistent).
Drug Interactions
The combination of meperidine and
monoamine oxidase inhibitors should be avoided as it may result in
hypertension, hypotension, hyperpyrexia, coma, or respiratory arrest. The cause
of this catastrophic interaction is incompletely understood. (The results of
failure to appreciate this drug interaction in the celebrated Libby Zion case
led to changes in work rules for house officers in the United States.)
Propofol, barbiturates, benzodiazepines,
and other central nervous system depressants can have synergistic
cardiovascular, respiratory, and sedative effects with opioids.
The biotransformation of alfentanil may
be impaired following treatment with erythromy-cin, leading to prolonged
sedation and respiratory depression.
Related Topics