Chapter: Modern Pharmacology with Clinical Applications: Pharmacokinetics
Additional Pharmacokinetic Parameters
ADDITIONAL
PHARMACOKINETIC PARAMETERS
Bioavailability
Bioavailability (designated
as F) is defined as the frac-tion of the administered drug reaching the
systemic cir-culation as intact drug. Bioavailability is highly depend-ent on
both the route of administration and the drug formulation. For example, drugs
that are given intra-venously exhibit a bioavailability of 1, since the entire
dose reaches the systemic circulation as intact drug. However, for other routes
of administration, this is not necessarily the case.
Subcutaneous, intramuscular,
oral, rectal, and other extravascular routes of administration require that the
drug be absorbed first, which can reduce bioavailability. The drug also may be
subject to metabolism prior to reaching the systemic circulation, again
potentially re-ducing bioavailability. For example, when the β-blocking agent
propranolol is given intravenously, F= 1, but when it is given orally, F= ~0.2,
suggesting that only ap-proximately 20% of the administered dose reaches the
systemic circulation as intact drug.
With respect to the effect of
drug formulation on bioavailability, the drug digoxin provides a good exam-ple.
Given orally as a solution, the bioavailability of digoxin approaches F= 1,
suggesting essentially com-plete bioavailability and one that approaches that
of the intravenous formulation. Digoxin liquid capsules also exhibit F =~1 when
given orally and thus are also com-pletely available. However, for digoxin
tablets, F =~0.7, suggesting incomplete bioavailability, probably because of
lack of absorption.
Two types of bioavailability can be calculated, de-pending on the formulations available and the informa-tion required. The gold standard is a calculation of the absolute bioavailability of a given product compared to the intravenous formulation (F =1). The absolute bioavailability of a drug can be calculated as:
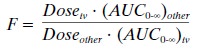
where the route of
administration is other than intra-venous (e.g., oral, rectal). For calculation
of absolute bioavailability, complete concentration-time profiles are needed
for both the intravenous and other routes of administration.
The other computation is that
of relative bioavail-ability. This
calculation is determined when two prod-ucts are compared to each other, not to
an intravenous standard. This is commonly calculated in the generic drug
industry to determine that the generic formulation (e.g., a tablet) is
bioequivalent to the original formula-tion (e.g., another tablet). Thus,
bioavailability is not routinely calculated in an individual patient but
re-served for product development by a drug manufac-turer. However, it is important
to have an idea of how formulations or routes of administration differ with
re-spect to bioavailability so as to allow proper dosage ad-justment when
changing formulations or routes of ad-ministration.
Clearance
Clearance is a
pharmacokinetic parameter used to de-scribe the efficiency of irreversible
elimination of drug from the body. More specifically, clearance is defined as
the volume of blood from which drug can be completely removed per unit of time
(e.g., 100 mL/minute). Clearance can involve both metabolism of drug to a
metabolite and excretion of drug from the body. For ex-ample, a molecule that
has undergone glucuronidation is described as having been cleared, even though
the molecule itself may not have left the body. Clearance of drug can be
accomplished by excretion of drug into the urine, gut contents, expired air,
sweat, and saliva as well as metabolic conversion to another form. However,
up-take of drug into tissues does not constitute clearance.
In the broadest sense, total
(systemic) clearance is the clearance of drug by all routes. Total (systemic)
clearance (Cl) can be calculated by either of the equa-tions given below:
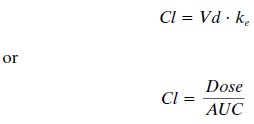
where Vd is the volume of
distribution and the remainder of the parameters
are as defined previ-ously. One must give the drug intravenously to assure 100%
bioavailability, because lack of 100% bioavail- ability can change the dose
numerator, which is re-quired to calculate total clearance. Frequently,
however, one wishes to calculate drug clearance but intravenous administration
is not feasible. In this situation, the ap-parent clearance (also called oral
clearance) can be es-timated by the following equation:
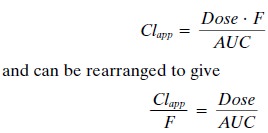
The term apparent clearance is used because the bioavailability of the
compound is unknown. Thus, esti-mations of apparent clearance will always be
higher than the true systemic clearance because of this un-known
bioavailability.
The final clearance value
that is frequently calcu-lated is that of renal clearance, or that portion of
clear-ance that is due to renal elimination. Renal clearance is calculated as:
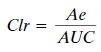
where Ae is the total amount
of drug excreted un-changed into the urine. Calculation of renal clearance is
especially useful for drugs that are eliminated primarily by the kidney.
Because clearance estimates
the efficiency of the body in eliminating drug, the calculation of clearance
can be especially useful in optimizing dosing of patients. Since this parameter
includes both the volume of distri-bution and the elimination rate, it adjusts
for differences in distribution characteristics and elimination rates among
people, thus permitting more accurate compar-isons among individuals. However,
as stated earlier, by far the easiest clearance parameter to estimate is that
of apparent (oral) clearance, since it does not require in-travenous
administration, yet this parameter can be pro-foundly affected by
bioavailability of the drug.
Volume of Distribution
Vd relates a concentration of
drug measured in the blood to the total amount of drug in the body. This
mathematically determined value gives a rough indica-tion of the overall
distribution of a drug in the body. For example, a drug with a Vd of
approximately 12 L (i.e., interstitial fluid plus plasma water) is probably
distrib-uted throughout extracellular fluid but is unable to pen-etrate cells.
In general, the greater the Vd, the greater the diffusibility of the drug.
The volume of distribution is
not an actual volume, since its estimation may result in a volume greater than
the volume available in the body (~40 L in a 70-kg adult). Such a value will
result if the compound is bound or sequestered at some extravascular site. For example, a highly lipid-soluble drug, such
as thiopental, that can be extensively stored in fat depots may have a Vd
con-siderably in excess of the entire fluid volume of the body. Thus, because
of their physicochemical character-istics, different drugs can have quite
different volumes of distribution in the same person.
The antiinflammatory drug
ibuprofen, for example, typically exhibits a volume of distribution of 0.14
L/kg such that for a 70-kg person, the Vd would be 10.8 L. This volume (10.8 L)
is approximately equal to the plasma volume of a person that size, suggesting
that this drug does not distribute widely into tissues (though it does reach
tissues to some degree to exert its action). In contrast, the antiarrhythmic
amiodarone has a Vd of 60 L/kg, giving a total Vd of 4200 L for this same 70-kg
per-son. This large Vd suggests that amiodarone distributes widely throughout
the body; in fact, it does distribute to various tissues, such as the liver,
lungs, eyes, and adipose tissue. Since the total volume of the body does not
equal 4200 L, it can clearly be seen that this is not a “real” vol-ume but one
that relates the blood concentration to the amount of drug in the body.
Protein Binding
Most drugs bind to plasma
proteins such as albumin and 1-acid glycoprotein (AGP) to some
degree. This be-comes clinically important as it is assumed that only un-bound
(free) drug is available for binding to receptors, being metabolized by
enzymes, and eliminated from the body. Thus, the free fraction of drug is
important. For example, phenytoin is approximately 90% bound to plasma
proteins, leaving 10% of the concentration in the blood as free drug and
available for pharmacologi-cal action and metabolism. If the presence of renal
dis-ease or a drug interaction were to alter the degree of protein binding to
only 80%, this change could have substantial clinical consequences. Even though
the total percent bound changes relatively little, the net result is to double
the amount of free drug. In fact, for pheny-toin, this can have clinical
consequences. However, for most drugs, displacement from protein binding sites
re-sults in only a transient increase in free drug concentra-tion, since the
drug is rapidly redistributed into other body water compartments. Thus, interactions
or changes in protein binding in most cases have little clinical effect despite
these theoretical considerations.
Related Topics