Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Genomics, Other “Omics” Technologies, Personalized Medicine, and Additional Biotechnology Related Techniques
Protein Engineering
Techniques To Modify And Study Proteins And Products Of Biotechnology
Protein Engineering
Many early biotechnology-produced protein drug candidates failed in clinical trials due to their short biological half-life, low affinity for their receptor, or immunogenicity (McCafferty and Glover, 2000). Recombinant DNA technology has made it possible to engineer specifically altered or new and novel protein molecules possessing tailored chemical and biological characteristics. Termed protein engineering, the deliberate design and construction of unique proteins with enhanced or novel molecular properties is a result of specifying the exact amino acid sequence (protein primary structure) of that protein (Narang, 1990; Richardson and Richardson, 1990; Cleland and Craik, 1996). When applied to enzymes, the process is often called enzyme engineering.
As described, the primary structure affects the protein’s conformation. The conformation of each and every amino acid component present in the protein influences the protein’s complex three-dimensional (3-D) structure. The conformational pre-ference of the protein chain residues determines the protein’s secondary structure including α-helices and β-sheets or reverse turns. The local secondary struc-tures are folded into 3-D tertiary structures made up of domains. The domains are not only structural units, but are also functional units often containing intact ligand binding (in a receptor) or enzyme catalytic sites. Thus, protein engineering provides an approach to modify a native protein’s structure specifically or to create a unique, new protein with a particular structure. Protein engineering has numerous power-ful theoretical and practical implications for examin-ing and modifying protein structure and function, probing enzyme mechanisms, investigating protein folding and conformation, enhancing protein stability, introducing detectable groups into proteins as an analytical tool, producing improved second genera-tion tailored biopharmaceuticals, and in the case of enzymes, improving catalytic function (Fothergill-Gilmore, 1993; Nixon et al., 1998).
Engineered proteins have been prepared by many different approaches. Direct chemical synthetic routes for small proteins with modified amino acid sequences have been devised using either solution chemistry or solid supports (chemistry occurring while reactants are attached to resin beads) techni-ques. Peptide synthesizers have been designed to automate the process. Dugas provides a useful overview of the chemistry of protein engineering (including site-directed mutagenesis) (Dugas, 1999). The synthesis of gene fragments coding for the mutation(s) is another approach to produce
engineered proteins (Johnson and Reitz, 1998). Completely synthetic genes of as many as 100 nucleotides coding for the desired mutation can be inserted into a gene of a prokaryotic (such as Phage M13) or eukaryotic expression vector. The resulting mutant gene (hybrid gene) is then cloned and expressed producing the engineered protein. The genetic route to engineered proteins is limited to the repertoire of the 20 natural amino acids, yet new technologies appear to be moving toward an ex-panded repertoire including moieties that would result in peptidomimetics. The purely chemical route allows for the introduction of alternative structures (e.g., non-natural amino acids) in the peptide chain (see section “Peptide Chemistry and Peptidomimetics”).
Site-Directed Mutagenesis
Site-directed mutagenesis (also called site-specific mutagenesis) is a protein engineering technique allowing specific amino acid residue (site-directed) alteration (mutation) to create new protein entities (Johnson and Reitz, 1998). Mutagenesis at a single amino acid position in an engineered protein is called a point mutation. Therefore, site-directed mutagenesis techniques can aid in the examination at the mole-cular level of the relationship between 3-D structure and function of interesting proteins.
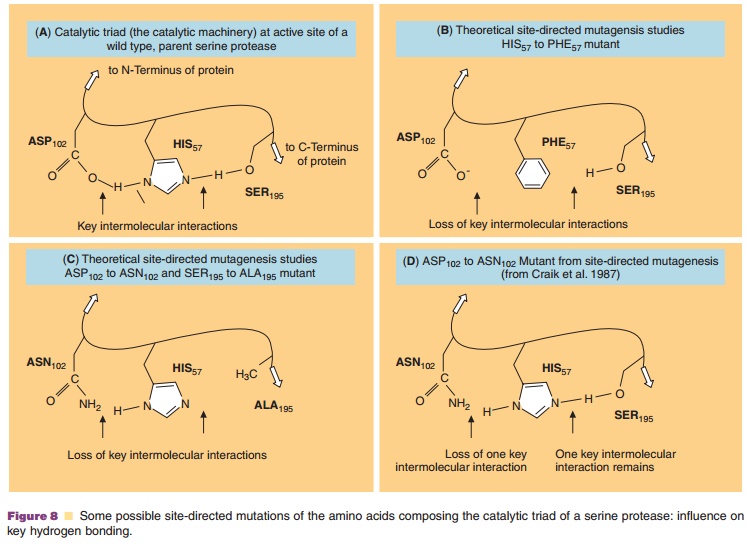
Figure 8 suggests an excellent example of possible theoretical mutations of the active site of a model serine protease enzyme that could be engineered to probe the mechanism of action of the enzyme. Structures B and C of Figure 8 represent a theoretical mutation to illustrate the technique. Craik and co-workers have actually tested the role of the aspartic acid residue in the serine protease catalytic triad Asp, His, and Ser. They replaced Asp102(carboxylate anion side chain) of trypsin with Asn (neutral amide side chain) by site-directed mutagenesis and observed a pH dependent change in the catalytic activity compared to the wild-type parent serine protease (Fig. 8, structure D) (Craik et al., 1987). Site-directed mutagenesis studies also provide invaluable insight into the nature of inter-molecular interactions of ligands with their receptors. For example, studies of the effect of the site-directed mutagenesis of various key amino acid residues on the binding of neurotransmitters to G-protein coupled receptors has helped define more accurate models for alpha-adrenergic, D2-dopaminergic, 5HT2a-serotoner-gic and both M1 and M3 muscarinic receptors (Bikker, et al., 1998).
Directed Evolution
Today, many of the techniques to engineer proteins with improved properties such as enhanced biological activity, improved catalytic specificity, metabolic stability, etc. are often referred to as “directed evolution” (Oelschlaeger and Mayo, 2005; Pelletier, 2007 and references contained therein). Studying the relationship between a protein’s sequence and the resulting protein’s property allows for a prediction of the optimal structure-property relationship and thus the “evolved” protein to be synthesized by standard techniques of biotechnology. Enzyme engineering to evolve a protein is achievedtypically by reasoned direct experimental manipulation of protein structure, computationally, or more recently, a combination of both (Fox et al., 2007). The variations resulting from all the possible protein sequences to be explored (re-ferred to as “sequence space”) to guide the directed evolution of even an average-sized protein are astronomically large. Thus, while the approach holds greattime.
Enzyme Engineering
Enzyme engineering is the application of protein engineering techniques to enzymatic molecules.Enzyme engineering can optimize catalytic reactions, improve an enzyme’s function under abnormal conditions, and enhance or change the catalytic reaction of unnatural substrates (Nixon, et al., 1998). An exciting application of protein engineering is the preparation of enzymes that have improved catalytic activity and stability in organic solvents, rather than requiring an aqueous environment. In that case, site-directed mutagenesis replaces hydrophilic, charged amino acids and hydrogen bonding residues at the surface of the enzyme with amino acids that stabilize the conformational stability of the protein at the organic solvent–protein surface interface.
The generation of enzyme hybrids, enzymes composed of elements of more than one enzyme, is an exciting area of current study using enzyme engineer-ing techniques. Some examples include the hybridiza-tion of the enzyme trypsin to hydrolyze either trypsin and chymotrypsin substrates, and the modification of the substrate specificity of lactate dehydrogenase (pyruvate) to include also oxaloacetate (Nixon, et al., 1998). While enzyme engineering is a powerful technique, it is difficult to engineer, via site-directedmutagenesis, a new catalytic function into an existing enzyme because of the precise spatial arrangement required for the catalytic functional groups at the active site. Fusion molecules (see below), however, are examples of protein-engineered products that may possess more than one activity or property. By fusing the secondary-structural elements or whole domains of enzymes, one could theoretically construct hybrid enzymes (or other proteins) capable of catalyzing reactions not observed in nature.
Fusion Proteins
Using ligation chemistry to fuse the gene-coding region for one protein with that of another protein, researchers have created chimeric proteins that combine the properties and activities of the two individual parents. The molecule created is called a fusion protein. Fusion proteins contain portions, or the entire amino acid sequences, of both parent proteins. Fusion proteins have found use in improv-ing the gene expression of a target protein, creating molecules with additive biological activities, and assessing the structure-activity relationships of re-gions in a protein important to its function.
Creating a fusion protein as an intermediate may facilitate gene expression of therapeutically useful proteins (or any protein). Human recombinant proin-sulin is expressed highly by cloning a fusion gene consisting of the codes for both proinsulin and the enzyme galactosidase. After recovering the fusion protein from E. coli culture, cleavage of the methio-nine peptide bond linking the two proteins with the chemical cyanogen bromide yields the free proinsulin.
Ligation chemistry can create DNA coding for fusion molecules with additive properties in compar-ison to the individual parent proteins. Numerous fusion proteins have been created that contain a toxin fused to another protein. Cutaneous T-cell lymphoma (CTCL) is a general term for a group of low-grade non-Hodgkin’s lymphomas affecting approximately 1000 new patients/year. For many patients, CTCL is a persistent, disfiguring and debilitating disease that requires multiple treatments over time. Malignant CTCL cells express one or more of the components of the IL-2 receptor. Thus, the IL-2 receptor may be a homing device to attract a “killer.” The IL-2 fusion protein DAB389 IL-2 (also called IL-2 fusion toxin) is a recombinant protein consisting of amino acid residues 2–133 of human IL-2 (the IL-2 residues replace the amino acids of the receptor-binding domain of the native diphtheria toxin) “fused” to the first 389 amino acid residues of diphtheria toxin (catalytic and lipophilic domains) (VanderSpek et al., 1993). Denileukin diftitox (Ontak) is such a FDA-approved rDNA-derived cytotoxic IL-2 “fusion” protein. The drug targets IL-2 receptors (the IL-2 portion), andbrings the diphtheria toxin directly to the cell to kill the CTCL targets. Studies have observed 30% of patients treated with Denileukin diftitox experience at least 50% reduction of tumor burden sustained for at least 6 weeks.
Many additional variations of diphtheria toxin-containing fusion proteins have been engineered including DAB389 CD4 (containing amino acids 1–178 contained in the V1 and V2 domains of human CD4; studied for the treatment of chronically HIV-infected cells), DAB389 IL-4 (linked to interleukin 4; treatment of myeloma and Kaposi’s sarcoma), DAB389 IL-6 (linked to interleukin 6; therapy of autoimmune diseases and cancer), DAB389 EGF (containing the amino acid sequence of epidermal growth factor; prevention of restenosis), and DAB389 hGM-CSF (fused peptide sequence of human GM-CSF; potential as an antileukemic agent).
Antibody Engineering
A pharmaceutically important application of protein engineering is the production of chimeras to examine the structure-activity relationships of a protein. An example is the engineering of humanized or fully human MAbs. These altered MAbs are prepared by expressing a chimeric antibody gene containing the code for both human and murine portions of the resulting antibody protein or the antibody gene for the fully human protein, respectively. The differences between species in the structure-activity relationships and the structure-function relationships of these chimeric or human antibodies can be examined by studying properties such as antigen specificity, affi-nity, and avidity (Pluckthun, 1992; Andersen and Reilly, 2004; Hoogenboom, 2005; Chowdhury and Wu, 2005). MAbs (chimeric, huma-nized, fully human) and improved antibody frag-ments are necessary to support research and pharmacotherapy. The application of protein engi-neering to the synthesis of new antibodies or improved antibody fragments is called antibody engineering. A significant effort is also invested in modifying glycosylation patterns and simplifying production methods.
Designed, potent antibody therapeutics consti-tutes one of the most rapidly growing class of human therapeutics (Carter, 2006). Immunoadhesins are anti-body engineered fusion proteins containing the immunoglobulin Fc effector domain and a molecule that will adhere specifically to other target molecules. Examples include the replacement of the variable region of an antibody with either the helper T-cell CD4 surface protein or tumor necrosis factor receptor (Fig. 9). These immunoadhesins would retain the antibody’s Fc effector region , but would display specificity for HIV or tumor necrosis
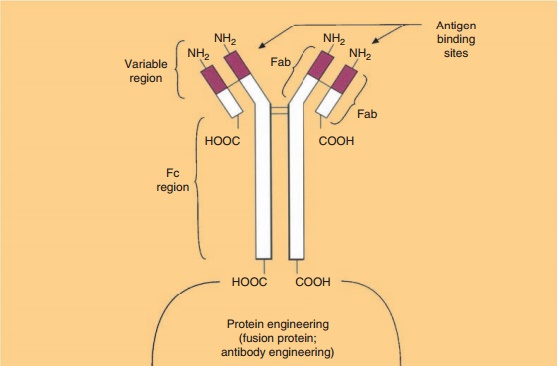
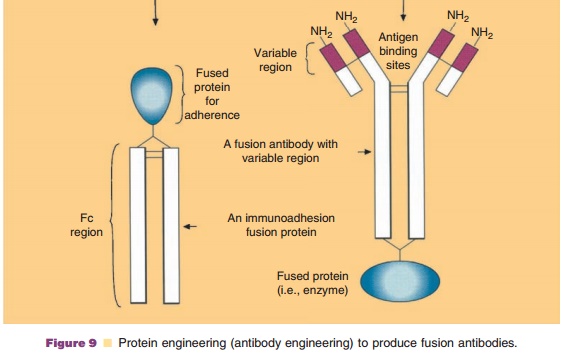
factor, respectively. Tumor necrosis factor-alpha is a proinflammatory cytokine released during various immune challenges. A soluble TNF-alpha receptor could bind to circulating TNF and remove the proinflammatory protein. The biopharmaceutical Etanercept (Enbrel) was approved for the reduction of signs and symptoms in rheumatoid arthritis patients refractory to disease-modifying antirheu-matic drugs. It is a fusion protein combining a rDNA, soluble P75 TNF receptor (this portion acts as natural antagonist to TNF) fused with a human IgG Fc region. Thus, the drug binds excess circulating TNF preventing it from binding to its membrane receptor.
Alternatively, a fused protein can be produced that consists of an antibody with an intact variable region to recognize and bind a specific target (i.e., an antifibrin antibody) along with an enzyme (i.e., tissue plasminogen activator, tPA) resulting in a more specific and potent agent (i.e., a fibrin specific thrombolytic agent). In other words, the antibody first attaches the enzyme (tPA) specifically to fibrin clots. The enzyme tPA will activate circulating plasminogen by converting it from plasminogen into plasmin. This plasmin locally attacks fibrin clots and dissolves them. The concept is called antibody-directed enzyme prodrug therapy, ADEPT.
Related Topics