Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Genomics, Other “Omics” Technologies, Personalized Medicine, and Additional Biotechnology Related Techniques
Genomics - An Introduction to “Omics” Technologies
Genomics
Sequencing the human genome and the genomes of other organisms has lead to an enhanced under-standing of human biology and disease. Many industry analysts predicted a tripling of pharmaceutical R&D productivity due to the sequencing of the human genome (Williams, 2007). Success to date has been limited and there is significant debate as to the impact of genomics on successful drug discovery and development (Nirmala, 2006; Caldwell et al., 2007). Validation of viable drug targets identified by genomics has been challenging (Bagowski, 2005). An interesting concept is that of the “druggable” genome (Hopkins and Groom, 2002). This is an estimate at 600 to 1,500 valid molecular targets for drug discovery, as assessed as the intersection of the number of human genes identified linked to disease with the subset of the human genome products that could be modulated by small-molecule targets. However, the genomics revolution has been the foundation for an explosion in “omics” technologies that find application in research to address neglected diseases.
Structural Genomics and the Human Genome Project
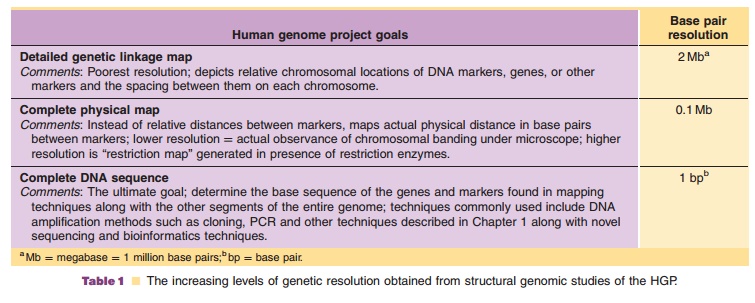
Genomics is the comparative study of the complete genome sequence and its function from different organisms (Caldwell et al., 2007). Initially, genetic analysis focused on structural genomics, basically the characterization of the structure of the genome. Structural genomics intersects the techniques of DNA sequencing, cloning, PCR, protein expression, crystallography, and data analysis. It focuses on the physical aspects of the genome through the construc-tion and analysis of gene sequences and gene maps. Proposed in the late 1980s, the publicly funded HGP or Human Genome Initiative (HGI) was officially sanctioned in October 1990 to map the structure and to sequence human DNA (Collins and Galas, 1993; Cantor and Smith, 1999). As described in Table 1, HGP structural genomics was envisioned to proceed through increasing levels of genetic resolution: de-tailed human genetic linkage maps [approximately 2 megabase pairs (Mb ¼ million base pairs) resolution], complete physical maps (0.1 Mb resolution), and ultimately complete DNA sequencing of the approxi-mately 3.5 billion base pairs (2, 3 pairs of chromo-somes) in a human cell nucleus [1 base pair (bp) resolution] (Griffiths et al., 2000). Projected for completion in 2003, the goal of the project was to learn not only what was contained in the genetic code, but also how to “mine” the genomic information to cure or help prevent the estimated 4,000 genetic diseases afflicting humankind. In May 1999, 700 million base pairs of the human genome were deposited in public archives. After merely fifteen months of additional studies, the figure had increased to greater than 4 billion base pairs (Pennisi, 2000). Two years earlier than projected, a milestone in genomic science was reached on June 26, 2000, whenresearchers at the privately funded Celera Genomics and the publicly funded Genome International Sequencing Consortium (the international collabora-tion associated with the HGP) jointly announced that they had completed sequencing 97% to 99% of the human genome. The journal Science rates the mapping of the human genome as its “breakthrough of the year” in its December 22, 2000 issue. The two groups published their results in 2001 (Venter et al., 2001; The Genome International Sequencing Consortium, 2001).
The genome sequencing strategies of the HGP and Celera Genomics differed (Brown, 2000). HGP utilized a “nested shotgun” approach. The human DNA sequence was “chopped” into segments of ever decreasing size and the segments put into rough order. Each segment was further “blasted” into small fragments. Each small fragment was sequenced and the sequenced fragments assembled according to their known relative order. The Celera researchers em-ployed a “whole shotgun” approach where they “blasted” the whole genome into small fragments. Each fragment was sequenced and assembled in order by identifying where they overlapped. Each approach required unprecedented computer resources.
Regardless of genome sequencing strategies, the collective results are impressive. More than 27 million high quality sequence reads provided five-fold cover-age of the entire human genome. Genomic studies have identified over 1 million single nucleotide polymorphisms (SNPs), binary elements of genetic variability. While original estimates of the number of human genes in the genome varied consistently between 80,000 to 120,000, the genome researchers unveiled a number far short of biologist’s predictions; 32,000 (Venter et al., 2001; The Genome International
Sequencing Consortium, 2001). Within months, others suggested that the human genome possesses between 65,000 and 75,000 genes (Wright et al., 2001). Approximately 25,000 genes is now most often cited number (Lee et al., 2006).
Newer structural genomics projects focus on lowering the average costs of structure determination while quantifying novel structures by direct sequence comparison (Chondonia and Brenner, 2006). The move toward low cost, high throughput sequencing is essential for the implementation of genomics in the individualized medicine clinical laboratory. The $1000 (U.S.) genome remains the target.
Functional and Comparative Genomics
Now that the sequencing of the entire genome is a reality, the chore of sorting through human, pathogen and other organism diversity factors and correlating them with genomic data to provide real pharmaceu-tical benefits is an active area of research (Kramer and Cohen, 2005). Pharmaceutical company drug pipe-lines are loaded with promising therapeutic agent leads, due in large part to the application of biotechnology to the discovery process. Early suppor-ters of the human genome project characterized it as a potential medical panacea that would rapidly add to the pipeline. However, attempts to translate genomic sequence data from structural genomics toward biological function has been predicted to fuel dis-coveries from the bench top to the patient’s bedside. This requires a through understanding of what genes do, how they are regulated, and the direct relationship between genes and their activity. The DNA sequence information itself rarely provides definitive informa-tion about the function and regulation of that particular gene. Yet there was hope that completing the human genome sequence would dramatically alter prevention, treatment, and even our definition of disease (Temple et al., 2001). The study of functional genomics is hoped to play a key role in new drug target identification and validation (Caldwell et al., 2007). After genome sequencing, this approach is the next step in the knowledge chain to identify functional gene products that are potential biotech drug leads and new drug discovery targets (Fig. 1). Functional genomics focuses on genome-wide patterns of gene expression, the mechanisms by which gene expression is coordinated, and the interrelation-ships of gene expression when a cellular environ-mental change occurs.
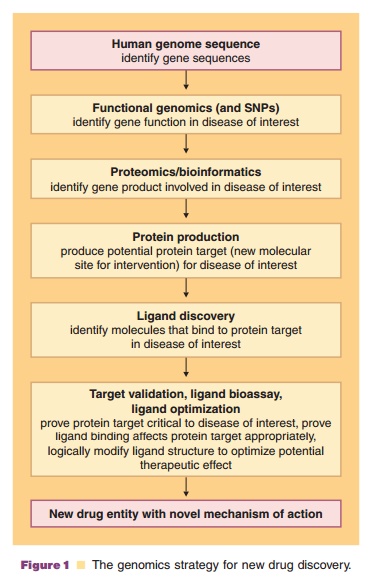
To relate functional genomics to therapeutic clinical outcomes, the human genome sequence must reveal the thousands of genetic variations among individuals that will become associated with diseases in the patient’s lifetime. Sequencing alone is not the end, simply the end of the beginning of the genomicmedicine era. Determining gene functionality in any organism opens the door for linking a disease to specific genes or proteins, which become targets for new drugs, methods to detect organisms (i.e., new diagnostic agents), and or biomarkers (the presence or change in gene expression profile that correlates with the risk, progression or susceptibility of a disease).
The face of biology has changed forever with the sequencing of the genomes of numerous organisms. Biotechnologies applied to the sequencing of the human genome are also being utilized to sequence the genomes of comparatively simple organisms as well as other mammals. Often, the proteins encoded by the genomes of lesser organisms and the regulation of those genes closely resemble the proteins and gene regulation in humans. Since model organisms are much easier to maintain in a laboratory setting, researchers are actively pursuing “comparative” genomics studies (Clark, 1999; Hardison, 2003). Unlocking genomic data for each of these organisms provides valuable insight into the molecular basis of inherited human disease (Karow, 2000). S. cerevisiae is a good model for studying cancer and is a common organism used in rDNA methodology. For example, it has become well known that women who inherit a gene mutation of the BRCA1 gene have a high risk, perhaps as high as 85%, of developing breast cancer before the age of 50 (source: www.ncbi.nlm.nih.gov). The first diagnostic product generated from genomic data was the BRCA1 test for breast cancer predisposition. The gene product of BRCA1 is a well-characterized protein implicated inboth breast and ovarian cancer. Evidence has accumu-lated suggesting that the Rad9 protein of S. cerevisiae is distantly, but significantly, related to the BRCA1 protein. The fruit fly possesses a gene similar to p53, the human tumor suppressor gene. Studying C. elegans has provided much of our early knowledge of apoptosis, the normal biological process of pro-grammed cell death. Greater than 90% of the proteins identified thus far from a common laboratory animal, the mouse, have structural similarities to known human proteins. Mapping the whole of a human cancer cell genome will pinpoint the genes involved in cancer and aid in the understanding of cell changes and treatment of human malignancies (Collins and Barker, 2007). The consensus coding sequences of human breast and colorectal cancers have been elucidated. The U.S. National Institutes of Health (NIH) Cancer Genome Atlas is one such project.
Not all comparative genomic studies are looking for similarities to the human genome. For example, some may provide the basis for creating new and novel potential antibiotic and antiviral targets for drug design (Guild, 1999). Comparative genomics is being used to provide a compilation of genes that code for proteins that are essential to the growth orviability of a pathogenic organism, yet differ from any human protein. The worldwide effort to sequence the severe acute respiratory syndrome (SARS) associated coronavirus genome is one such example that will aid diagnosis, antiviral discovery, and vaccine develop-ment (Marra et al., 2003). Assuring selective toxicity to the organism, not the human patient, this genomic mining of new targets for drug design may aid the quest for new antibiotics in a clinical environment of increasing incidence of antibiotic resistance.
DNA banking, the collection, storage, and analysis of hundreds of thousands of specimens containing analyzable DNA, is proving to be a valuable tool for genetics research (Thornton et al., 2005). All nucleated cells, including cells from blood, hair follicles, buccal swabs, cancer biopsies, and urine specimens, are suitable specimens for DNA analysis in the present or at a later date. These banks have become valuable resources for the discovery of new and improved medical treatments in several disease states, especially cancer.
Epigenomics
Using a whole-genome approach, epigenomics is the study of environmental or developmental genetic effects such as DNA methylation, on gene function. Thus, epigenomics focuses on those genes whose function is determined by external factors. Epidemiological evidence increasingly suggests that external factors including environmental exposures early in development have a role in disease susceptibility (Jirtle and Skinner, 2007).
Related Topics