Chapter: Pathology: Principles of Neoplasia
Carcinogenesis
CARCINOGENESIS
Carcinogenesis
is a multistep process, and development of all human
cancersappears to require the accumulation of multiple genetic changes. These
changes can involve either inherited germline mutations or acquired mutations.
Once a sin-gle severely mutated cell forms, monoclonal expansion of the cell’s
line can cause a tumor. Most important mutations in tumorogenesis involve
growth promoting genes (proto-oncogenes), growth inhibiting tumor suppressor
genes, or the genes regulating apoptosis and senescence.
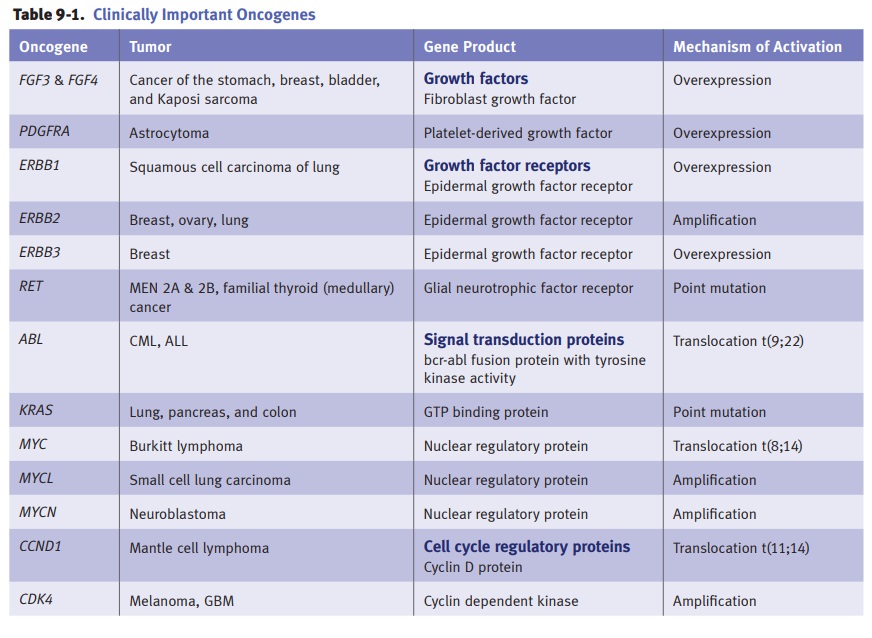
Activation
of growth promoting oncogenes.Proto-oncogenes are normal
cellulargenes involved with growth and cellular differentiation. Oncogenes are
derived from proto-oncogenes by either a change in the gene sequence, resulting
in a new gene product (oncoprotein), or a loss of gene regulation resulting in
overexpression of the normal gene product. Mechanisms of oncogene activation
include point muta-tions, chromosomal translocations, gene amplification, and
insertional mutagenesis. Activated oncogenes lack regulatory control and are
overexpressed, resulting in unregulated cellular proliferation.
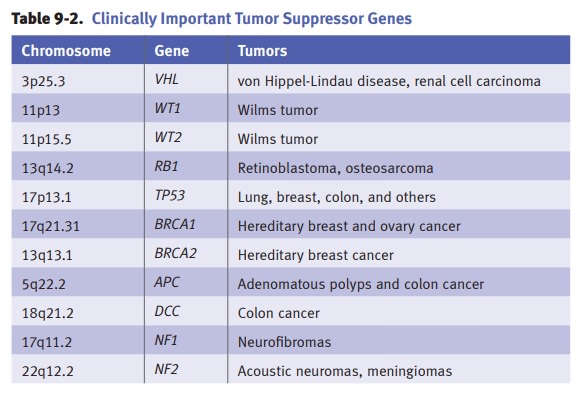
Inactivation
of tumor suppressor genes.Tumor suppressor genes encode
proteins thatregulate and suppress cell proliferation by inhibiting progression
of the cell through the cell cycle. The mechanism of action of tumor suppressor
genes may vary. As exam-ples, p53 prevents a cell with damaged DNA from
entering S-phase, while Rb prevents the cell from entering S-phase until the
appropriate growth signals are present.
Knudson’s “two hit hypothesis” states
that at least 2 tumor suppressor genesmust be inactivated for oncogenesis. In
cancers arising in individuals with inherited germline mutations, the “first
hit” is the inherited germline mutation and the “second hit” is an acquired
somatic mutation. Examples of inherited germline mutations include familial
retinoblastoma (in which germline muta-tion of RB1 on chromosome 13 is associated with a high rate of
retinoblastoma and osteosarcoma) and Li-Fraumini syndrome (in which germline
mutation of TP53 on chromosome 17 is
associated with a high rate of many types of tumors).
Regulation
of apoptosis.Tumor genesis related to changes in regulation of
apotosisoccurs in the follicular lymphomas that have the translocation
t(14;18). Normally, Bcl-2 prevents apoptosis (programmed cell death). In the
follicular lymphomas with this translocation, the Bcl-2 regulator of apoptosis
is overexpressed, because the translocation connects the immunoglobulin heavy
chain gene on chromosome 14 (which turns on easily in B lymphocytes) to the BCL2 gene on chromosome 18, thereby
leading to a situation in which lymphocytes fail to die as expected and instead
produce a tumor.
Other
examples of apoptosis regulators include Bax, Bad, bcl-xS, and Bid; p53
pro-motes apoptosis in mutated cells by stimulating bax synthesis. The protein
c-myc promotes cellular proliferation and when associated with p53 leads to
apoptosis and when associated with Bcl-2 inhibits apoptosis.
Limitless
replication is possible due in part to upregulation of telomerase.
Sustained
angiogenesis is possible due in part to activation of the Notch
signalingpathway.
Invasiveness/metastasis.
Malignant cells must dissociate from tumors (loss ofE-cadherin function) and
degrade the extracellular matrix before spreading to dis-tant sites.
Cancer-associated glycans are being investigated for their role in cancer
spread and as targets for therapy.