Chapter: Modern Pharmacology with Clinical Applications: The Renin–Angiotensin– Aldosterone System and Other Vasoactive Substances
Antagonists of the Renin–Angiotensin System
ANTAGONISTS OF
THE RENIN–ANGIOTENSIN SYSTEM
A summary of the agents that
inhibit the renin– angiotensin system and their sites of action is provided in
Figure 18.3.
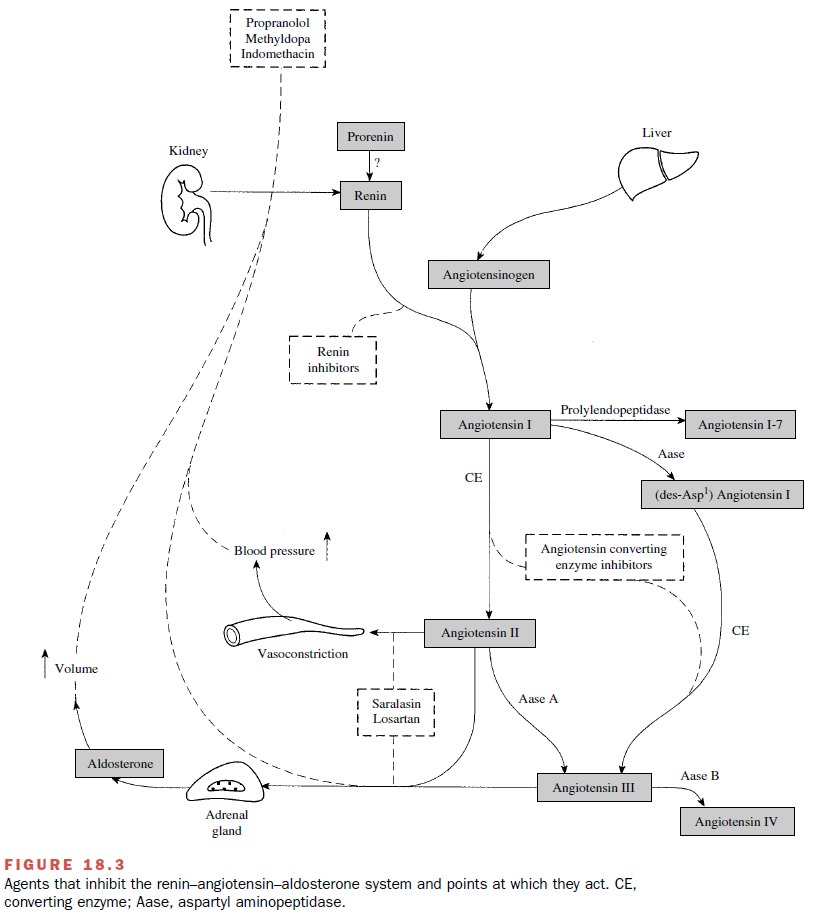
Renin Inhibitors
The acid protease inhibitor pepstatin and some ana-logues of
angiotensinogen can competitively inhibit the formation of angiotensin I by
human renin. Highly spe-cific renin inhibitors may prove beneficial as
antihyper-tensive agents or in the treatment of congestive heart failure.
Despite extensive efforts to develop renin in-hibitors, most compounds capable
of inhibiting renin are large peptidelike molecules that lack adequate physical
chemical properties to permit oral absorption.
Angiotensin-Converting Enzyme Inhibitors
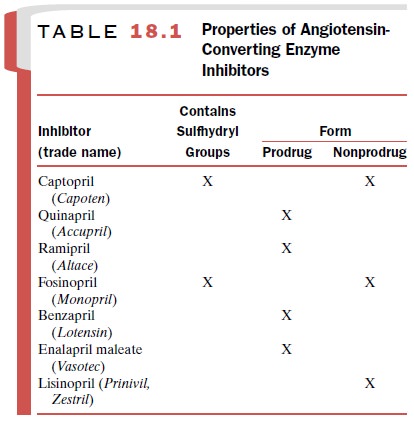
Many of the orally active ACE
inhibitors are prodrugs. These include perindopril, quinapril, benazepril,
ramipril, enalapril, trandolapril, and fosinopril.
Captopril
Captopril (Capoten) is an orally effective ACE
inhibitor with a sulfhydryl moiety that is used in binding to the active site
of the enzyme. Captopril blocks the blood pressure responses caused by the
administration of an-giotensin I and decreases plasma and tissue levels of
an-giotensin II.
Pharmacological Actions
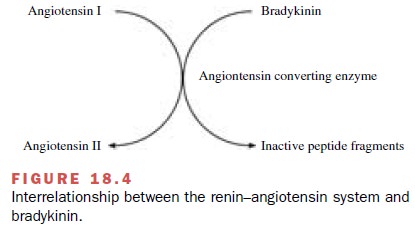
Treatment with captopril reduces blood pressure in patients with renovascular disease and in patients with essential hypertension. The decrease in arterial pressure is related to a reduction in total peripheral resistance. Most studies demonstrate a good correlation between the hypotensive effect of inhibitors and the degree of blockade of the renin–angiotensin system. Many of the pharmacological effects of captopril are attributable to the inhibition of angiotensin II synthesis. However, ACE is a relatively nonselective enzyme that also ca-tabolizes a family of kinins to inactive products (Fig. 18.4). Bradykinin, one of the major kinins, acts as a va-sodilator through mechanisms related to the production of nitric oxide and prostacyclin by the vascular en-dothelium. Thus, administration of the ACE inhibitor captopril not only inhibits angiotensin II production but also prevents the breakdown of bradykinin. Increases in bradykinin concentrations after administration of ACE inhibitors contribute to the therapeutic efficacy of these compounds in the treatment of hypertension and con-gestive heart failure. However, alterations in bradykinin concentrations are also thought to contribute to cough and angioedema sometimes seen after ACE inhibition.
The hypotensive response to
captopril is accompa-nied by a fall in plasma aldosterone and angiotensin II
levels and an increase in plasma renin activity. Serum potassium levels are not
affected unless potassium sup-plements or potassium-sparing diuretics are used
con-comitantly; this can result in severe hyperkalemia.
There is no
baroreflex-associated increase in heart rate, cardiac output, or myocardial
contractility in response to the decrease in pressure, presumably because
capto-pril decreases the sensitivity of the baroreceptor reflex.
Captopril enhances cardiac output in patients with congestive heart failure by inducing a reduction in ven-tricular afterload and preload. Converting enzyme in-hibitors have been shown to decrease the mass and wall thickness of the left ventricle in both normal and hy-pertrophied myocardium. ACE inhibitors lack meta-bolic side effects and do not alter serum lipids.
Pharmacokinetics
The onset of action following
oral administration of captopril is about 15 minutes, with peak blood levels
achieved in 30 to 60 minutes. Its apparent biological half-life is
approximately 2 hours, with its antihyperten-sive effects observed for 6 to 10
hours. The kidneys ap-pear to play a major role in the inactivation of
captopril.
Clinical Uses
Captopril, as well as other
ACE inhibitors, is indi-cated in the treatment of hypertension, congestive
heart failure, left ventricular dysfunction after a myocardial infarction, and
diabetic nephropathy. In the treatment of essential hypertension, captopril is
considered first-choice therapy, either alone or in combination with a thiazide
diuretic. Decreases in blood pressure are pri-marily attributed to decreased
total peripheral resist-ance or afterload. An advantage of combining captopril
therapy with a conventional thiazide diuretic is that the thiazide-induced
hypokalemia is minimized in the pres-ence of ACE inhibition, since there is a
marked de-crease in angiotensin II–induced aldosterone release.
If the patient is
asymptomatic, captopril can be used as monotherapy in the treatment of
congestive heart failure. The use of ACE inhibitors in the treatment of
congestive heart failure is supported by results from large-scale clinical
trials demonstrating a general reduc-tion in the relative risk of death. In
symptomatic pa-tients captopril should be used in conjunction with a di-uretic
because of the weak natriuretic properties of ACE inhibitors. In combination,
captopril will reduce afterload and preload and prevent diuretic-induced
ac-tivation of the renin–angiotensin system. Finally, ACE inhibitors may slow
the progression of congestive heart failure by limiting left ventricular
hypertrophy.
In the treatment of diabetic
nephropathy associated with type I insulin-dependent diabetes mellitus,
capto-pril decreases the rate of progression of renal insuffi-ciency and
retards the worsening of renal function.
Adverse Actions
Approximately 10% of the
patients treated with captopril report a dose-related maculopapular rash that
often disappears when the dosage of captopril is reduced. Other common adverse
effects are fever, a persistent dry cough (incidence as high as 39%), initial
dose hypotension, and a loss of taste that may result in anorexia. These
effects are reversed when drug ther-apy is discontinued. More serious
toxicities include a 1% incidence of proteinuria and glomerulonephritis; less
common are leukopenia and agranulocytosis. Since food reduces the
bioavailability of captopril by 30 to 40%, administration of the drug an hour
before meals is recommended. All converting enzyme in-hibitors are
contraindicated in patients with bilateral renal artery disease or with unilateral
renal artery dis-ease and one kidney. Use under these circumstances may result
in renal failure or paradoxical malignant hypertension.
Prodrug Angiotensin-converting Enzyme Inhibitors
Most orally effective
inhibitors of peptidyl dipeptide hydrolase are prodrug ester compounds that
must be hydrolyzed in plasma to the active moiety before be-coming effective.
These drugs include benazepril (Lotensin),
enalapril (Vasotec), fosinopril (Monopril), moexipril (Univasc),
quinapril (Accupril), perindopril (Aceon), and ramipril (Altace). The ester group pro-motes
absorption of the compound from the gastroin-testinal tract. In contrast to
captopril, the recommended dosing interval for these prodrug compounds is once
to twice daily. These compounds are otherwise generally similar to captopril in
their mechanism of action and in-dicated uses.
All prodrug ACE inhibitors
are indicated for use as first-choice agents in the treatment of hypertension
and congestive heart failure. In addition, results from clini-cal trials
demonstrate that ramipril, a prodrug ACE in-hibitor, can reduce the rate of
death, myocardial infarc-tion, and stroke in a broad range of high-risk
patients who did not have heart failure. These results suggest that ACE
inhibitors may be useful in the management of ischemia and atherosclerosis.
While essentially all ACE
inhibitors have a similar mechanism of action and therefore exhibit similar
effi-cacy in the treatment of hypertension and congestive heart failure, these
drugs differ slightly in their phar-macokinetic profiles. Enalapril,
lisinopril, and quinapril are excreted primarily by the kidney, with minimal
liver metabolism, while the other prodrug compounds are metabolized by the
liver and renally excreted. Thus, in patients with renal insufficiency, the
half-life of renally excreted ACE inhibitors is pro-longed. In addition,
patients with impaired liver func- tion may have a compromised ability to
convert pro-drug to the active drug moiety, so the efficacy of the compounds
may be reduced. In addition, compounds dependent on liver metabolism for
elimination may exhibit an increase in plasma half-life. An additional property
that distinguishes among these prodrugs is their individual abilities to bind
tightly to tissue ACE, as opposed to the circulating form of the enzyme. Of the
prodrug inhibitors, quinapril and perindopril bind most tightly.
All ACE inhibitors are
contraindicated during preg-nancy. Their administration to pregnant women
during the second and third trimesters of pregnancy has been associated with
fetal and neonatal injury, including fetal death. A summary of the ACE
inhibitors and their properties is provided in Table 18.1.
Angiotensin Receptor Antagonists
Angiotensin II can bind with
high affinity to two distinct receptors, termed the angiotensin type 1 (AT1)
and the angiotensin type 2 (AT2) receptor. These receptors be-long to a
superfamily of G protein–coupled receptors that contain seven transmembrane
regions. The amino acid sequence of these receptors is highly conserved across
species. The AT1 and AT2 receptors share only 34% homology, have distinct
signal transduction path-ways, and are not necessarily found on the same cell
type or tissue. As previously discussed, most of the phys-iological effects of
angiotensin II are mediated through effects at the AT1 receptor.
Mechanism of Action and Pharmacological Actions
Losartan (Cozaar) was the first imidazole AT1
receptor antagonist developed and is a selective competitive antagonist to
angiotensin II. Other AT1 receptor antag-onists approved for the treatment of
hypertension in-clude valsartan (Diovan),
irbesartan (Avapro), can-desartan
cilexetil (Atacand), telmisartan (Micardis), and eprosartan (Teveten). These antagonists share some
pharmacological characteristics, including a high affinity for the AT1
receptor, little to no affinity for the AT2 re-ceptor, high protein binding,
and the ability to produce an almost insurmountable blockade of the AT1
recep-tor. Although all of the AT1 receptor antagonists are competitive
blocking drugs, they only slowly dissociate from the receptor, and their
effects cannot be easily overcome.
The administration of an AT1
receptor antagonist results in a decrease in total peripheral resistance
(af-terload) and cardiac venous return (preload). All of the physiological
effects of angiotensin II, including stimu-lation of the release of
aldosterone, are antagonized in the presence of an AT1 receptor antagonist.
Reductions in blood pressure occur independently of the status of the
renin–angiotensin system, making these drugs effec-tive antihypertensives even
in patients with normal to low activity of the renin–angiotensin system.
Following the chronic administration of an AT1 receptor antago-nist, plasma
renin activity increases as a result of re-moval of the angiotensin II negative
feedback.
Pharmacokinetic Profiles of AT1 Receptor Antagonists
While all AT1 receptor
antagonists share the same mechanism of action, they differ in their
pharmacoki-netic profiles. Losartan is well absorbed following oral administration
and undergoes significant first-pass liver metabolism to an active metabolite,
EXP3174. This metabolite is a long-acting (6–8 hours) noncompetitive antagonist
at the AT1 receptor that contributes to the pharmacological effects of
losartan. Production of the long-acting metabolite accounts for the sustained
anti-hypertensive properties of losartan following chronic therapy, which would
otherwise eventually be over-whelmed by removal of the negative feedback system
(inhibition of renin release) for angiotensin II produc-tion. Following oral
administration, 6% of losartan is excreted unchanged in the urine.
Valsartan has a higher
affinity for the AT1 receptor than losartan, does not have an active
metabolite, and has a slightly longer duration of action than losartan.
Irbesartan exhibits high bioavailability and high affinity for the AT1
receptor, does not have an active metabo-lite, and has a considerably longer
duration of action than losartan. Candesartan cilexetil has an active metabolite
with a long duration of action, is a prodrug, and exhibits an AT1 receptor
affinity 80 times that of losartan. Telmisartan is the longest-acting AT1
receptor antagonist and has no active metabolites. In contrast, eprosartan has
the shortest half-life of the AT1 receptor antagonists and has been suggested
to exhibit selective blockade of some effects of angiotensin II more than
others.
Clinical Uses of AT1 Receptor Antagonists
Angiotensin type 1 receptor
antagonists are effective as monotherapy in the treatment of hypertension.
While all of the AT1 receptor antagonists are effective in the treatment of
hypertension, several comparative studies have suggested that longer-acting AT1
receptor antago-nists, such as irbesartan, candesartan, and telmisartan, may be
more effective than the shorter-acting antago-nists at providing 24-hour
control of blood pressure. In large-scale clinical trials, AT1 receptor
antagonists did not exhibit a clear advantage over ACE inhibitors in re-ducing
morbidity and mortality from congestive heart failure. Therefore, the use of
AT1 receptor antagonists in the treatment of congestive heart failure is
generally re-stricted to patients who do not tolerate ACE inhibitors.
Adverse Effects of AT1 Receptor Antagonists
All AT1 receptor antagonists have
adverse effects that are not significantly different from those of a placebo,
although first-dose hypotension may occur. Unlike ACE inhibitors, AT1 receptor
antagonists do not pro-duce a cough, suggesting that this side effect may be
re-lated to the buildup of bradykinin levels that occurs as a result of
converting enzyme inhibition rather than to a reduction in angiotensin II
levels. An additional dif-ference between AT1 receptor antagonists and ACE
in-hibitors is that angiotensin II is capable of interacting at the AT2
receptor in patients treated with an AT1 re-ceptor antagonist (but not
following inhibition of ACE). The clinical significance of this difference is
not understood.
Finally, additional enzymes
have been identified that are capable of forming angiotensin II from
angiotensin I, suggesting that inhibition of ACE may not be sufficient for the
total elimination of angiotensin II. In contrast, AT1 receptor antagonists are
capable of blocking the ef-fects of angiotensin II regardless of its enzymatic
route of formation. All AT1 receptor antagonists, like the ACE inhibitors, are
contraindicated during pregnancy.
Related Topics