Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Genomics, Other “Omics” Technologies, Personalized Medicine, and Additional Biotechnology Related Techniques
Screening and Synthesis - Biotechnology and Drug Discovery
Screening and Synthesis
Traditionally, drug discovery programs relied heavily upon random screening followed by analog synthesis and lead optimization via structure-activity relationshipstudies. Discovery of novel, efficacious, and safer small molecule medicinal agents with appropriate drug-like characteristics is an increasingly costly and complex process (Arlington, 2000, Williams, 2007). Therefore, any method allowing for a reduction in time and money is extremely valuable. Advances in biotechnology have contributed to a greater understanding of the cause and progression of disease and have identified new ther-apeutic targets forming the basis of novel drug screens. New technical discoveries in the fields of proteomics for target discovery and validation, and systems biology are expected to facilitate the discovery of new agents with novel mechanisms of action for diseases that were previously difficult or impossible to treat. In an effort to decrease the cost of identifying and optimizing useful, quality drug leads against a pharmaceutically important target; researchers have developed newer approaches including high-throughput screening and high-throughput synthesis methods.
Advances in Screening: High Throughput Screening (HTS)
Recombinant DNA technology has provided the ability to clone, express, isolate and purify receptor enzymes, membrane bound proteins, and other binding proteins in larger quantities than ever before. Instead of using receptors present in animal tissues or partially purified enzymes for screening, in vitro bioassays now utilize the exact human protein target. Applications of biotechnology to in vitro screening include the improved preparation of: (1) cloned membrane-bound receptors expressed in cell-lines carrying few endogenous receptors; (2) immobilized preparations of receptors, antibodies and other ligand-binding proteins; and (3) soluble enzymes and extracellular cell-surface expressed protein recep-tors. In most cases today, biotechnology contributes directly to the understanding, identification and/or the generation of the drug target being screened (e.g., radioligand binding displacement from a cloned protein receptor).
Previously, libraries of synthetic compounds along with natural products from microbial fermenta-tion, plant extracts, marine organisms and inverte-brates provide a diversity of molecular structures that were screened randomly. Screening can be made more directed if the compounds to be investigated are selected on the basis of structural information about the receptor or natural ligand. The development of sensitive radioligand binding assays and the access to fully automated, robotic screening techniques have accelerated the screening process.
High-throughput screening (HTS) provides for the bioassay of thousands of compounds in multiple assays at the same time (Houston and Banks, 2003; Cik and Jurzak, 2007). The process is automated with robots and utilizes multi-well microtiter plates. While 96-well microtiter plates are a versatile standard in HTS, the development of 1536-and 3456-well nano-plate formats and enhanced robotics brings greater miniaturization and speed to cell based and biochem-ical assays. Now, companies can conduct 100,000 bioassays a day. In addition, modern drug discovery and lead optimization with DNA microarrays allows researchers to track hundreds to thousands of genes.
Enzyme inhibition assays and radioligand bind-ing assays are the most common biochemical tests employed. The technology has become so sophisti-cated and the interactive nature of biochemical events so much better understood (through approaches such as system biology) that HT whole cell assays have become commonplace. Reporter gene assays are routinely utilized in HTS (Ullmann, 2007). Typically, a reporter gene, that is a reporter that indicates the presence or absence of a particular gene product that in tern reflects the changes in a biological process or pathway, is transfected into a desired cell. When the gene product is expressed in the living cell, the reporter gene is transcribed and the reporter is translated to yield a protein that is measured biochemically. A common reporter gene codes for the enzyme luciferase, and the intensity of the resulting green fluorescent protein (i.e., a quantitative measure of concentration) is a direct function of the assayed molecule’s ability to stimulate or inhibit the biologic process or signaling pathway understudy. A further advance in HT screening technologies for lead optimization is rapid, high content pharma-cology. This HT screening approach can be used to evaluate solubility, adsorption, toxicity, metabolism, etc. (Brown et al., 2003).
High-Throughput Chemistry: Combinatorial Chemistry and Multiple Parallel Synthesis
Traditionally, small drug molecules were synthesized by joining together structural pieces in a set sequence to prepare one product. One of the most powerful tools to optimize drug discovery is automated high-throughput synthesis. When conducted in a combinatorial approach, high-throughput synthesis provides for the simultaneous preparation of hun-dreds or thousands of related drug candidates (Fenniri, 2000; Sucholeiki, 2001; Mason and Pickett, 2003; Pirrung, 2004). The molecular libraries gener-ated are screened in high-throughput screening assays for the desired activity, and the most active molecules are identified and isolated for further development.
There are two overall approaches to high-throughput synthesis (Bunin et al., 2003; Mitscher and Dutta, 2003; Seneci, 2007; Ashton and Moloney, 2007). True combinatorial chemistry applies methods to substantially reduce the number of synthetic operations or steps needed to synthesize large numbers of compounds. Combichem, as it is some-times referred to, is conducted on solid supports (resins) to facilitate the manipulations required to reduce labor. Differing from combinatorial chemistry,multiple parallel synthesis procedures apply automa-tion to the synthetic process, but the number of operations needed to carry out a synthesis is practically the same as the conventional approach. Thus, the potential productivity of multiple parallel methods is not as high as combinatorial chemistries. Parallel chemistries can be conducted on solid-phase supports or in solution. Figure 13 provides an illustration of a combinatorial mix-and-match process in which a simple building block (a starting material such as an amino acid, peptide, heterocycle, other small molecule, etc.) is joined to one or more other simple building blocks in every possible combination.
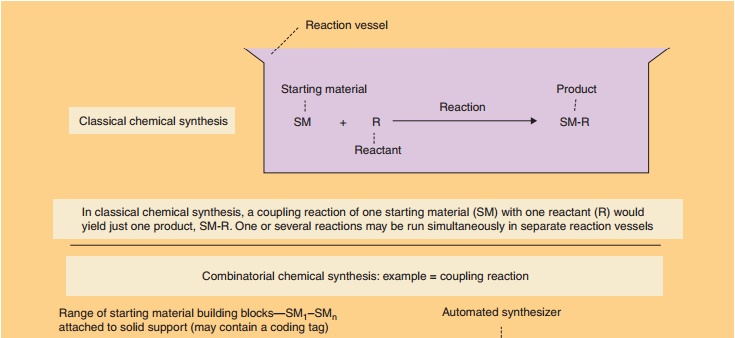
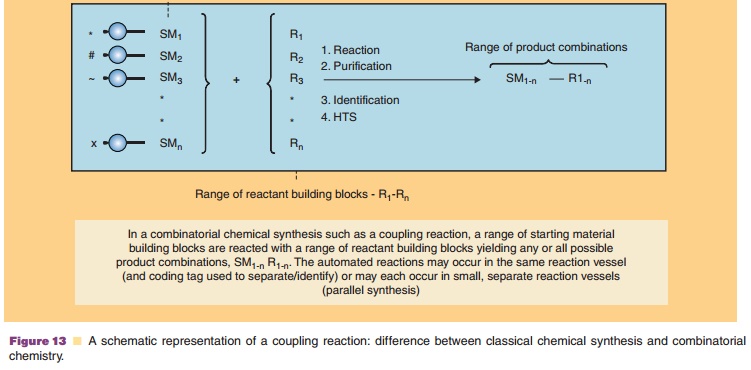
Assigning the task to automated synthesizing equip-ment results in the rapid creation of large collections or libraries (as large as 10,000 compounds) of diverse molecules. Ingenious methods have been devised to direct the molecules to be synthesized, to identify the structure of the products, to purify the products via automation, and to isolate compounds. When coupled with high-throughput screening, thousands of com-pounds can be generated, screened, and evaluated for further development in a matter of weeks.
Building blocks include amino acids, peptides, nucleotides, carbohydrates, lipids, and a diversity of small molecule scaffolds or templates (Mason and

Related Topics