Chapter: Medical Physiology: Dominant Role of the Kidney in Long-Term Regulation of Arterial Pressure and in Hypertension: The Integrated System for Pressure Control
The Renin-Angiotensin System: Its Role in Pressure Control and in Hypertension
The Renin-Angiotensin System: Its Role in Pressure Control and in Hypertension
Aside from the capability of the kidneys to control arterial pressure through changes in extracellular fluid volume, the kidneys also have another powerful mechanism for controlling pressure. It is the renin-angiotensin system.
Renin is a protein enzyme released by the kidneyswhen the arterial pressure falls too low. In turn, it raises the arterial pressure in several ways, thus helping to correct the initial fall in pressure.
Components of the Renin-Angiotensin System
Figure 19–9 shows the functional steps by which the renin-angiotensin system helps to regulate arterial pressure.
Renin is synthesized and stored in an inactive form called prorenin in the juxtaglomerular cells (JG cells) of the kidneys. The JG cells are modified smooth muscle cells located in the walls of the afferent
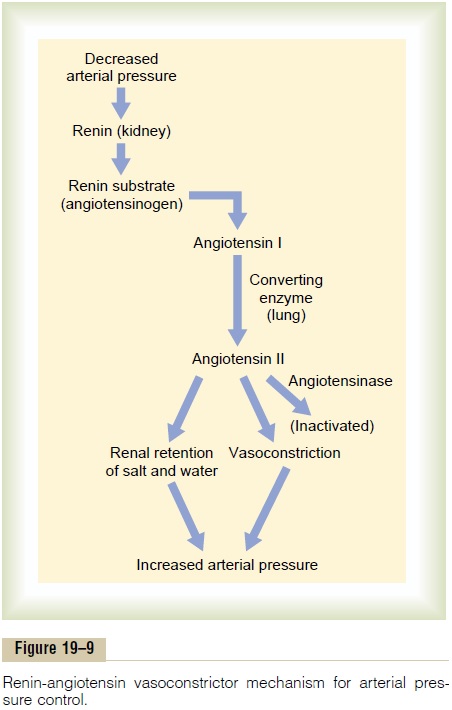
arterioles immediately proximal to the glomeruli. Whenthe arterial pressure falls, intrinsic reactions in the kidneys themselves cause many of the prorenin mole-cules in the JG cells to split and release renin. Most of the renin enters the renal blood and then passes out of the kidneys to circulate throughout the entire body. However, small amounts of the renin do remain in the local fluids of the kidney and initiate several intrarenal functions.
Renin itself is an enzyme, not a vasoactive sub-stance. As shown in the schema of Figure 19–9, renin acts enzymatically on another plasma protein, a globulin called renin substrate (or angiotensinogen), to release a 10-amino acid peptide, angiotensin I. Angiotensin I has mild vasoconstrictor properties but not enough to cause significant changes in circulatory function. The renin persists in the blood for 30 minutes to 1 hour and continues to cause formation of still more angiotensin I during this entire time.
Within a few seconds to minutes after formation of angiotensin I, two additional amino acids are split from the angiotensin I to form the 8-amino acid peptide angiotensin II. This conversion occurs almost entirely in the lungs while the blood flows through the small vessels of the lungs, catalyzed by an enzyme called converting enzyme that is present in the endothelium of the lung vessels.
Angiotensin II is an extremely powerful vasocon-strictor, and it also affects circulatory function in other ways as well. However, it persists in the blood only for 1 or 2 minutes because it is rapidly inactivated by multiple blood and tissue enzymes collectively called angiotensinases.
During its persistence in the blood, angiotensin II has two principal effects that can elevate arterial pressure. The first of these, vasoconstriction in manyareas of the body, occurs rapidly. Vasoconstrictionoccurs intensely in the arterioles and much less so in the veins. Constriction of the arterioles increases the total peripheral resistance, thereby raising the arterial pressure, as demonstrated at the bottom of the schema in Figure 19–9. Also, the mild constriction of the veins promotes increased venous return of blood to the heart, thereby helping the heart pump against the increasing pressure.
The second principal means by which angiotensin increases the arterial pressure is to decrease excretionof both salt and water by the kidneys. This slowlyincreases the extracellular fluid volume, which then increases the arterial pressure during subsequent hours and days. This long-term effect, acting through the extracellular fluid volume mechanism, is even more powerful than the acute vasoconstrictor mecha-nism in eventually raising the arterial pressure.
Rapidity and Intensity of the Vasoconstrictor Pressure Response to the Renin-Angiotensin System
Figure 19–10 shows a typical experiment demon-strating the effect of hemorrhage on the arterial pressure under two separate conditions: (1) with the renin-angiotensin system functioning and (2) without
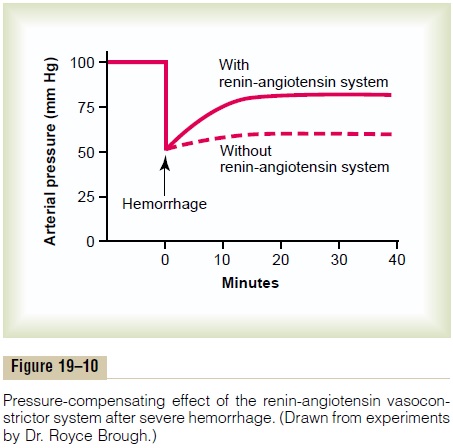
the system functioning (the system was interrupted by a renin-blocking antibody). Note that after hemor-rhage—enough to cause acute decrease of the arterial pressure to 50 mm Hg—the arterial pressure rose back to 83 mm Hg when the renin-angiotensin system was functional. Conversely, it rose to only 60 mm Hg when the renin-angiotensin system was blocked. This shows that the renin-angiotensin system is powerful enough to return the arterial pressure at least halfway back to normal within a few minutes after severe hemorrhage. Therefore, sometimes it can be of lifesaving service to the body, especially in circulatory shock.
Note also that the renin-angiotensin vasocon-strictor system requires about 20 minutes to become fully active. Therefore, it is somewhat slower to act for pressure control than are the nervous reflexes and the sympathetic norepinephrine-epinephrine system.
Effect of Angiotensin in the Kidneys to Cause Renal Retention of Salt and Water— An Especially Important Means for Long-Term Control of Arterial Pressure
Angiotensin causes the kidneys to retain both salt and water in two major ways:
1. Angiotensin acts directly on the kidneys to cause salt and water retention.
2.Angiotensin causes the adrenal glands to secrete aldosterone, and the aldosterone in turn increases salt and water reabsorption by the kidney tubules.
Thus, whenever excess amounts of angiotensin cir-culate in the blood, the entire long-term renal–body fluid mechanism for arterial pressure control auto-matically becomes set to a higher arterial pressure level than normal.
Mechanisms of the Direct Renal Effects of Angiotensin to Cause Renal Retention of Salt and Water. Angiotensin has severaldirect renal effects that make the kidneys retain salt and water. One major effect is to constrict the renal arterioles, thereby diminishing blood flow through the kidneys. As a result, less fluid filters through the glomeruli into the tubules. Also, the slow flow of blood reduces the pressure in the peritubular capillaries, which causes rapid reabsorption of fluid from the tubules. And still a third effect is that angiotensin has important direct actions on the tubular cells them-selves to increase tubular reabsorption of sodium and water. The total result of all these effects is significant, sometimes decreasing urine output less than one fifth of normal.
Stimulation of Aldosterone Secretion by Angiotensin, and the Effect of Aldosterone in Increasing Salt and Water Retention by the Kidneys. Angiotensin is also one of the mostpowerful stimulators of aldosterone secretion by the adrenal glands, as we shall discuss in relation to body fluid regulation and in relation to adrenal gland function. Therefore, when the renin-angiotensin system becomes activated, the rate of aldosterone secretion usually also increases; and an important subsequent function of aldosterone is to cause marked increase in sodium reabsorption by the kidney tubules, thus increasing the total body extracellular fluid sodium. This increased sodium then causes water retention, as already explained, increas-ing the extracellular fluid volume and leading second-arily to still more long-term elevation of the arterial pressure.
Thus both the direct effect of angiotensin on the kidney and its effect acting through aldosterone are important in long-term arterial pressure control. However, research in our own laboratory has sug-gested that the direct effect of angiotensin on the kidneys is perhaps three or more times as potent as the indirect effect acting through aldosterone—even though the indirect effect is the one most widely known.
Quantitative Analysis of Arterial Pressure Changes Caused by Angiotensin. Figure 19–11 shows a quantitative analysisof the effect of angiotensin in arterial pressure control. This figure shows two renal output curves as well as a line depicting normal level of sodium intake. The left-hand renal output curve is that measured in dogs whose renin-angiotensin system had been blocked by the drug captopril (which blocks the conversion of angiotensin Ito angiotensin II, the active form of angiotensin). The right-hand curve was measured in dogs infused contin-uously with angiotensin II at a level about 2.5 times the normal rate of angiotensin formation in the blood. Note the shift of the renal output curve toward higher pres-sure levels under the influence of angiotensin II. This shift is caused by both the direct effects of angiotensin on the kidney and the indirect effect acting through aldosterone secretion, as explained above.
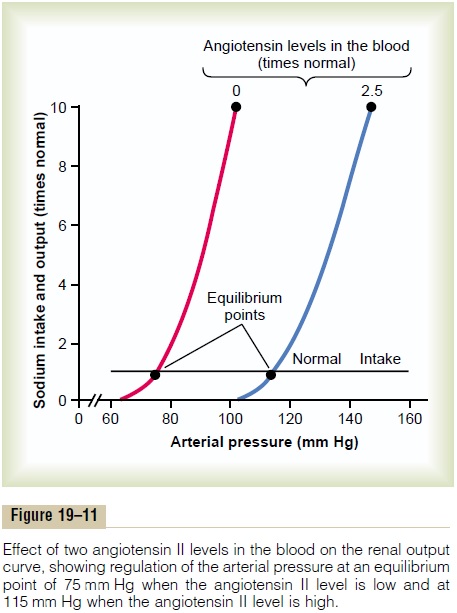
Finally, note the two equilibrium points, one for zero angiotensin showing an arterial pressure level of 75 mm Hg, and one for elevated angiotensin showing a pressure level of 115 mm Hg. Therefore, the effect of
Role of the Renin-Angiotensin System in Maintaining a Normal Arterial Pressure Despite Wide Variations in Salt Intake
One of the most important functions of the renin-angiotensin system is to allow a person to eat either very small or very large amounts of salt without causing great changes in either extracellular fluid volume or arterial pressure. This function is explained by the schema in Figure 19–12, which shows that the initial effect of increased salt intake is to elevate the extracellular fluid volume and this in turn to elevate the arterial pressure. Then, the increased arterial pres-sure causes increased blood flow through the kidneys, which reduces the rate of secretion of renin to a much lower level and leads sequentially to decreased renal retention of salt and water, return of the extracellular fluid volume almost to normal, and, finally, return of the arterial pressure also almost to normal. Thus, the renin-angiotensin system is an automatic feedback mechanism that helps maintain the arterial pressure at or near the normal level even when salt intake is increased. Or, when salt intake is decreased below normal, exactly opposite effects take place.
To emphasize the efficacy of the renin-angiotensin system in controlling arterial pressure, when the system functions normally, the pressure rises no more increase in salt intake. Conversely, when the renin- angiotensin system is blocked, the same increase in salt intake sometimes causes the pressure to rise 10 times the normal increase, often as much as 50 to 60 mm Hg.

Types of Hypertension in Which Angiotensin Is Involved: Hypertension Caused by a Renin-Secreting Tumor or by Infusion of Angiotensin II
Occasionally a tumor of the renin-secreting juxta-glomerular cells (the JG cells) occurs and secretes tremendous quantities of renin; in turn, equally large quantities of angiotensin II are formed. In all patients in whom this has occurred, severe hypertension has developed. Also, when large amounts of angiotensin are infused continuously for days or weeks into animals, similar severe long-term hypertension develops.
We have already noted that angiotensin can increase the arterial pressure in two ways:
1.By constricting the arterioles throughout the entire body, thereby increasing the total peripheral resistance and arterial pressure; this effect occurs within seconds after one begins to infuse angiotensin.
2.By causing the kidneys to retain salt and water; over a period of days, this, too, causes hypertension and is the principal cause of the long-term continuation of the elevated pressure.
“One-Kidney” Goldblatt Hypertension. When one kidney isremoved and a constrictor is placed on the renal artery of the remaining kidney, as shown in Figure 19–13, the immediate effect is greatly reduced pressure in the renal artery beyond the constrictor, as demonstrated by the dashed curve in the figure. Then, within seconds or minutes, the systemic arterial pressure begins to rise and continues to rise for several days. The pressure usually rises rapidly for the first hour or so, and this is followed by a slower additional rise during the next several days. When the systemic arterial pressure reaches its new stable pressure level, the renal arterial pressure (the dashed curve in the figure) will have returned almost all the way back to normal.The hyper-tension produced in this way is called “one-kidney”Goldblatt hypertension in honor of Dr. Goldblatt, whofirst studied the important quantitative features of hypertension caused by renal artery constriction.
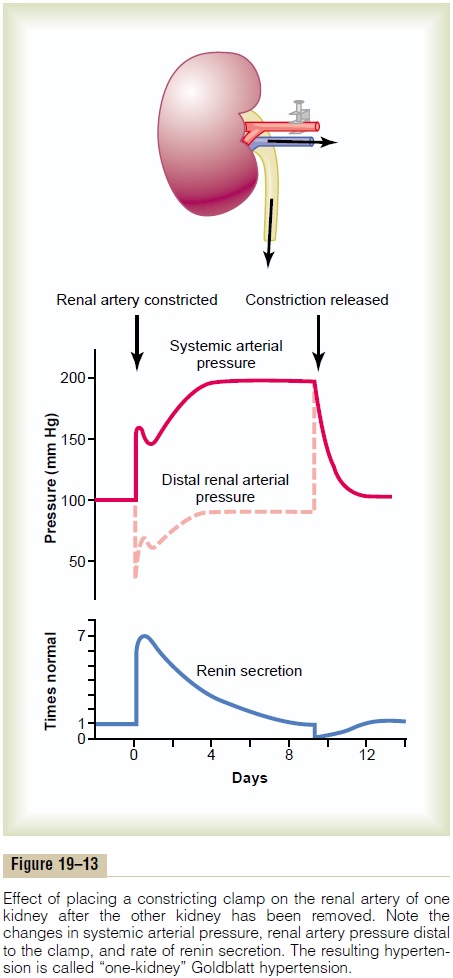
The early rise in arterial pressure in Goldblatt hypertension is caused by the renin-angiotensin vaso-constrictor mechanism. That is, because of poor blood flow through the kidney after acute constriction of the renal artery, large quantities of renin are secreted by the kidney, as demonstrated by the lowermost curve in Figure 19–13, and this causes increased angiotensin II and aldosterone in the blood. The angiotensin in turn raises the arterial pressure acutely. The secretion of renin rises to a peak in an hour or so but returns nearly to normal in 5 to 7 days because the renal arte-rial pressure by that time has also risen back to normal, so that the kidney is no longer ischemic.
The second rise in arterial pressure is caused by retention of salt and water by the constricted kidney (that is also stimulated by angiotensin II and aldos-terone). In 5 to 7 days, the body fluid volume will have increased enough to raise the arterial pressure to its new sustained level. The quantitative value of this sus-tained pressure level is determined by the degree of constriction of the renal artery. That is, the aortic pres-sure must rise high enough so that renal arterial pres-sure distal to the constrictor is enough to cause normal urine output.
“Two-Kidney” Goldblatt Hypertension. Hypertension alsocan result when the artery to only one kidney is con-stricted while the artery to the other kidney is normal. This hypertension results from the following mecha-nism: The constricted kidney secretes renin and also retains salt and water because of decreased renal arte-rial pressure in this kidney. Then the “normal” oppo-site kidney retains salt and water because of the renin produced by the ischemic kidney. This renin causes formation of angiotension II and aldosterone both of which circulate to the opposite kidney and cause it also to retain salt and water. Thus, both kidneys, but for dif-ferent reasons, become salt and water retainers. Con-sequently, hypertension develops.
Hypertension Caused by Diseased Kidneys That Secrete Renin Chronically. Often, patchy areas of one or both kidneysare diseased and become ischemic because of local vascular constrictions, whereas other areas of the kidneys are normal. When this occurs, almost identical effects occur as in the two-kidney type of Goldblatt hypertension. That is, the patchy ischemic kidney tissue secretes renin, and this in turn, acting through the formation of angiotensin II, causes the remaining kidney mass also to retain salt and water. Indeed, one of the most common causes of renal hypertension, especially in older persons, is such patchy ischemic kidney disease.
Other Types of Hypertension Caused by Combinations of Volume Loading and Vasoconstriction
Hypertension in the Upper Part of the Body Caused by Coarctation of the Aorta. One out of every few thousand babies isborn with pathological constriction or blockage of the aorta at a point beyond the aortic arterial branches to the head and arms but proximal to the renal arteries, a condition called coarctation of the aorta. When this occurs, blood flow to the lower body is carried by mul-tiple, small collateral arteries in the body wall, with much vascular resistance between the upper aorta and the lower aorta. As a consequence, the arterial pressure in the upper part of the may be 40-50 per cent higher than that in the lower body.
The mechanism of this upper-body hypertension is almost identical to that of one-kidney Goldblatt hyper-tension.That is, when a constrictor is placed on the aorta above the renal arteries, the blood pressure in both kidneys at first falls, renin is secreted, angiotensin and aldosterone are formed, and hypertension occurs in the upper body. The arterial pressure in the lower body at the level of the kidneys rises approximately to normal, but high pressure persists in the upper body. The kidneys are no longer ischemic, so that secretion of renin and formation of angiotensin and aldosterone return to normal. Likewise, in coarctation of the aorta, the arterial pressure in the lower body is usually almost normal, whereas the pressure in the upper body is far higher than normal.
Role of Autoregulation in the Hypertension Caused by Aortic Coarctation. A significant feature of hypertension causedby aortic coarctation is that blood flow in the arms, where the pressure may be 40 to 60 per cent above normal, is almost exactly normal. Also, blood flow in the legs, where the pressure is not elevated, is almost exactly normal. How could this be, with the pressure in the upper body 40 to 60 per cent greater than in the lower body? The answer is not that there are differences in vasoconstrictor substances in the blood of the upper and lower body, because the same blood flows to both areas. Likewise, the nervous system innervates both areas of the circulation similarly, so that there is no reason to believe that there is a difference in nervous control of the blood vessels. The only reasonable answer is that long-term autoregulation develops so nearly completely that the local blood flow control mechanisms have com-pensated almost 100 per cent for the differences in pres-sure. The result is that, in both the high-pressure area and the low-pressure area, the local blood flow is con-trolled almost exactly in accord with the needs of the tissue and not in accord with the level of the pressure. One of the reasons these observations are so important is that they demonstrate how nearly complete the long-term autoregulation process can be.
Hypertension in Preeclampsia (Toxemia of Pregnancy). Approx-imately 5 to 10 per cent of expectant mothers develop a syndrome called preeclampsia (also called toxemia ofpregnancy). One of the manifestations of preeclampsiais hypertension that usually subsides after delivery of the baby. Although the precise causes of preeclampsia are not completely understood, ischemia of the placenta and subsequent release by the placenta of toxic factors are believed to play a role in causing many of the man-ifestations of this disorder, including hypertension in the mother. Substances released by the ischemic placenta, in turn, cause dysfunction of vascular endothelial cells throughout the body, including the blood vessels of the kidneys.This endothelial dysfunction decreases release ofnitric oxide and other vasodilator substances, causingvasoconstriction, decreased rate of fluid filtration from the glomeruli into the renal tubules, impaired renal-pressure natriuresis, and development of hypertension.
Another pathological abnormality that may con-tribute to hypertension in preeclampsia is thickening of the kidney glomerular membranes (perhaps caused by an autoimmune process), which also reduces the rate of glomerular fluid filtration. For obvious reasons, the arte-rial pressure level required to cause normal formation of urine becomes elevated, and the long-term level of arterial pressure becomes correspondingly elevated. These patients are especially prone to extra degrees of hypertension when they have excess salt intake.
Neurogenic Hypertension. Acute neurogenic hypertensioncan be caused by strong stimulation of the sympatheticnervous system. For instance, when a person becomesexcited for any reason or at times during states of anxiety, the sympathetic system becomes excessively stimulated, peripheral vasoconstriction occurs every-where in the body, and acute hypertension ensues.
Acute Neurogenic Hypertension Caused by Sectioning the Barore- ceptor Nerves. Another type ofacuteneurogenic hyper-tension occurs when the nerves leading from the baroreceptors are cut or when the tractus solitarius is destroyed in each side of the medulla oblongata (these are the areas where the nerves from the carotid and aortic baroreceptors connect in the brain stem). The sudden cessation of normal nerve signals from the baroreceptors has the same effect on the nervous pressure control mechanisms as a sudden reduction of the arterial pressure in the aorta and carotid arteries. That is, loss of the normal inhibitory effect on the vasomotor center caused by normal baroreceptor nervous signals allows the vasomotor center suddenly to become extremely active and the mean arterial pres-sure to increase from 100 mm Hg to as high as 160 mm Hg. The pressure returns to nearly normal within about 2 days because the response of the vasomotor center to the absent baroreceptor signal fades away, which is called central “resetting” of the baroreceptor pressure control mechanism. Therefore, the neurogenic hyper-tension caused by sectioning the baroreceptor nerves is mainly an acute type of hypertension, not a chronic type.
Spontaneous Hereditary Hypertension in Lower Animals. Spon-taneous hereditary hypertension has been observed in a number of strains of lower animals, including several different strains of rats, at least one strain of rabbits, and at least one strain of dogs. In the strain of rats that has been studied to the greatest extent, the Okamoto strain, there is evidence that in early development of the hypertension, the sympathetic nervous system is con-siderably more active than in normal rats. However, in the late stages of this type of hypertension, two struc-tural changes have been observed in the nephrons of the kidneys: (1) increased preglomerular renal arterial resistance and (2) decreased permeability of the glomerular membranes. These structural changes could easily be the basis for the long-term continuance of the hypertension. In other strains of hypertensive rats, impaired renal function also has been observed.
“Primary (Essential) Hypertension”
About 90 to 95 per cent of all people who have hyper-tension are said to have “primary hypertension,” also widely known as “essential hypertension” by many cli-nicians. These terms mean simply that the hypertensionis of unknown origin, in contrast to those forms ofhypertension that are secondary to known causes, such as renal artery stenosis. In some patients with primary hypertension, there is a strong hereditary tendency, the same as occurs in animal strains of genetic hyperten-sion discussed above.
In most patients, excess weight gain and sedentarylifestyle appear to play a major role in causing hyper-tension. The majority of patients with hypertension are overweight, and studies of different populations suggest that excess weight gain and obesity may account for as much as 65 to 70 percent of the risk for developing primary hypertension. Clinical studies have clearly shown the value of weight loss for reduc-ing blood pressure in most patients with hypertension. In fact, new clinical guidelines for treating hyper-tension recommend increased physical activity and weight loss as a first step in treating most patients with hypertension.
Some of the characteristics of primary hypertension caused by excess weight gain and obesity include:
1. Cardiac output is increased due, in part, to theadditional blood flow required for the extra adipose tissue. However, blood flow in the heart, kidneys, gastrointestinal tract, and skeletal muscle also increases with weight gain due to increased metabolic rate and growth of the organs and tissues in response to their increased metabolic demands. As the hypertension is sustained for many months and years, total peripheral vascular resistance may be increased.
2. Sympathetic nerve activity, especially in the kidneys, is increased in overweight patients. The causes ofincreased sympathetic activity in obesity are not fully understood, but recent studies suggest that hormones, such as leptin, released from fat cells may directly stimulate multiple regions of the hypothalamus, which, in turn, have an excitatory influence on the vasomotor centers of the brain medulla.
3. Angiotensin II and aldosterone levels are increased two- to threefold in many obese patients. This maybe caused partly by increased sympathetic nerve stimulation, which increases renin release by the kidneys and therefore formation of angiotensin II, which, in turn, stimulates the adrenal gland to secrete aldosterone.
4. The renal-pressure natriuresis mechanism is impaired, and the kidneys will not excrete adequate amounts of salt and water unless the arterial pressure is high or unless kidney function is somehow improved. In other words, if the meanarterial pressure in the essential hypertensive person is 150 mm Hg, acute reduction of the mean arterial pressure artificially to the normal value of 100 mm Hg (but without otherwise altering renal function except for the decreased pressure) will cause almost total anuria, and the person will retain salt and water until the pressure rises back to the elevated value of 150 mm Hg. Chronic reductions in arterial pressure with effective antihypertensive therapies, however, usually do not cause marked salt and water retention by the kidneys because these therapies also improve renal-pressure natriuresis, as discussed below.
Experimental studies in obese animals and obese patients suggest that impaired renal-pressure natri-uresis in obesity hypertension is caused mainly by increased renal tubular reabsorption of salt and water due to increased sympathetic nerve activity and increased levels of angiotensin II and aldosterone. However, if hypertension is not effectively treated, there may also be vascular damage in the kidneys that can reduce the glomerular filtration rate and increase the severity of the hypertension. Eventually uncon-trolled hypertension associated with obesity can lead to severe vascular injury and complete loss of kidney function.
Graphical Analysis of Arterial Pressure Control in Essential Hypertension. Figure 19–14 is a graphical analysis ofessential hypertension. The curves of this figure are called sodium-loading renal function curvesbecause the arterial pressure in each instance is increased very slowly, over many days or weeks, by gradually
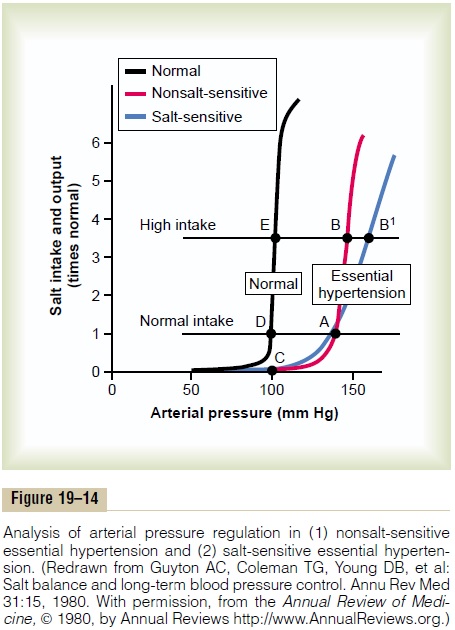
increasing the level of sodium intake. The sodium-loading type of curve can be determined by increasing the level of sodium intake to a new level every few days, then waiting for the renal output of sodium to come into balance with the intake, and at the same time recording the changes in arterial pressure.
When this procedure is used in essential hyperten-sive patients, two types of curves, shown to the right in Figure 19–14, can be recorded in essential hyperten-sive patients, one called (1) nonsalt-sensitivehyperten-sion and the other (2) salt-sensitive hypertension. Note in both instances that the curves are shifted to the right, to a much higher pressure level than for normal people. Now, let us plot on this same graph (1) a normal level of salt intake and (2) a high level of salt intake representing 3.5 times the normal intake. In the case of the person with nonsalt-sensitive essential hypertension, the arterial pressure does not increase significantly when changing from normal salt intake to high salt intake. Conversely, in those patients who have salt-sensitive essential hypertension, the high salt intake significantly exacerbates the hypertension.
Two additional points should be emphasized: (1) Salt-sensitivity of blood pressure is not an all-or-none characteristic—it is a quantitative characteristic, with some individuals being more salt-sensitive than others.
(2) Salt-sensitivity of blood pressure is not a fixed characteristic; instead, blood pressure usually becomes more salt-sensitive as a person ages, especially after 50 or 60 years of age.
The reason for the difference between nonsalt-sensitive essential hypertension and salt-sensitive hypertension is presumably related to structural or functional differences in the kidneys of these two types of hypertensive patients. For example, salt-sensitive hypertension may occur with different types of chronic renal disease due to gradual loss of the functional units of the kidneys (the nephrons) or to normal aging. Abnormal function of the renin-angiotensin system can also cause blood pres-sure to become salt-sensitive.
Treatment of Essential Hypertension. Current guidelinesfor treating hypertension recommend, as a first step, lifestyle modifications that are aimed at increasing physical activity and weight loss in most patients. Unfortunately, many patients are unable to lose weight, and pharmacological treatment with antihy-pertensive drugs must be initiated.
Two general classes of drugs are used to treat hyper-tension: (1) vasodilator drugs that increase renal blood flow and (2) natriuretic or diuretic drugs that decrease tubular reabsorption of salt and water.
Vasodilator drugs usually cause vasodilation in many other tissues of the body as well as in the kidneys. Different ones act in one of the following ways: (1) by inhibiting sympathetic nervous signals to the kidneys or by blocking the action of the sympa-thetic transmitter substance on the renal vasculature, (2) by directly relaxing the smooth muscle of the renal vasculature, or (3) by blocking the action of the renin-angiotensin system on the renal vasculature or renal tubules.
Those drugs that reduce reabsorption of salt and water by the renal tubules include especially drugs that block active transport of sodium through the tubular wall; this blockage in turn also prevents the reabsorption of water..
Related Topics