Chapter: Biochemical Pharmacology : Introduction
Synthetic drugs may exceed the corresponding physiological agonists in selectivity
Synthetic drugs may exceed
the corresponding physiological agonists in selectivity
Angiotensin is an example of
a peptide hormone. Peptide hormones and neurotransmitters are very numerous,
and new ones are constantly being discovered, as are new loca-tions and
receptors for known ones. While several drugs exist that act on peptide
receptors (most notably, opioids), drug development generally lags behind the
physiological characterization. The situation is quite different with an-other
group of hormones / transmitters, which are small-er molecules, most of them
related to amino acids. With many of these, the availability of drugs has
enabled the characterization of different classes of receptors and their
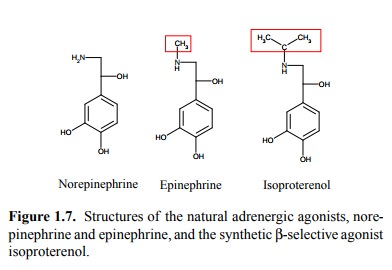
physiological roles. The classical example is the distinction ofα- and β-adrenergic receptors (which we will consider
in more detail later on in this course). While both epinephrine and
norepinephrine act on either receptor (though with somewhat different potency),
the distinction became very clear with the synthetic analog isoproterenol,
which acts very strongly on β-receptors but is virtually inactive on α-receptors (Figure 1.7).
Agonists and antagonists that
are more selective than the physiological mediators are both theoretically
interesting and of great practical importance. As a clinically signifi-cant
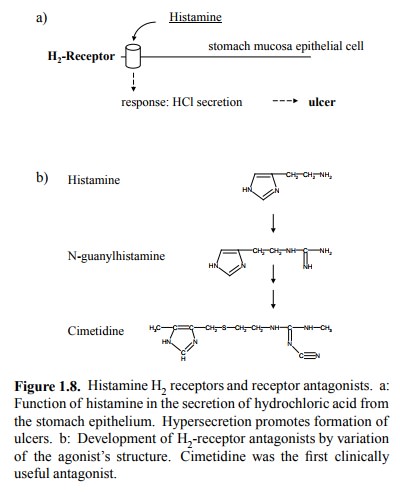
example of a selective receptor antagonist, we may consider the H2
histamine receptor in the stomach, which is involved in the secretion of
hydrochloric acid (Figure 1.8a). The mediator itself – histamine – was used as
starting point in the search for analogs that would bind to the receptor but not
activate it. The first derivative that displayed strongly reduced stimulatory
activity (while still binding to the re-ceptor, of course) was
N-guanylhistamine (Figure 1.8b). Further structural modification yielded
cimetidine, which was the first clinically useful H 2 receptor
blocker. It rep-resented a major improvement in ulcer therapy at the time and
is still in use today, although more modern drugs have largely taken its place.
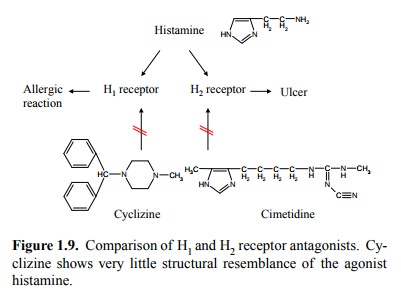
While H2-selective
blockers retain some structural resem-blance to the original mediator
(histamine), the same cannot be said of the likewise clinically useful H1
blockers, which were developed for the treatment of allergic diseases such as
hay fever (Figure 1.9).
Indeed, the H1
blockers do seem to be plagued by signifi-cant `cross-talk' to receptors other
than histamine receptors. This is not uncommon – many agents, particularly
those that readily penetrate into the central nervous system, have incompletely
defined receptor specificities, although they are usually given a label
suggesting otherwise. They are frequently used regardless on a empirical basis,
often for fairly diverse indications.
Related Topics