Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Gene Therapy
Regulation and Oversight of Gene Therapy Products
REGULATION AND OVERSIGHT OF GENE THERAPY PRODUCTS
Experimental use of gene medicines is initiated only after careful
review of the approach and trials monitored to protect the patient. The process
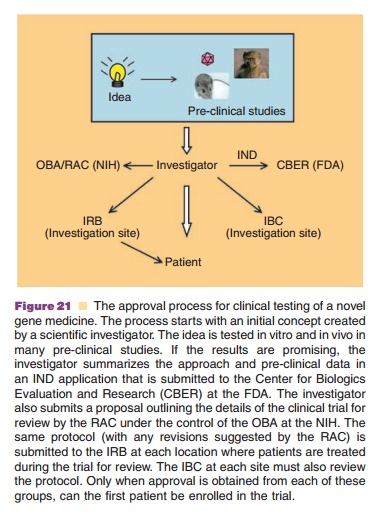
to obtain approval for clinical testing from regulatory agencies in the United
States is summarized in Figure 20. The Center for Biologics Evaluation and
Research (CBER) of the FDA reviews gene therapy Investigational New Drug (IND)
applications sub-mitted by an investigator or sponsor. The National Institutes
of Health (NIH) establishes guidelines for genetic research and implements
policies through thescientific concern about the ethical, scientific and safety
aspects of genetic research. Scientific and ethical review by the RAC is
mandatory for clinical trials occurring at or sponsored by institutions
receiving NIH funding for recombinant DNA research. Other protocols not meeting
these criteria may also be submitted voluntarily for RAC review. The Office for
Human Research Protections (OHRP) is responsible for monitoring and
implementing compliance and supplying educational materials protecting human
research subjects. The Institutional Biosafety Committee (IBC) and the
Institutional Review Board (IRB) at each investigative site must also review
and approve protocols and consent forms for any gene therapy clinical trial.
Enrollment of patients for the trial begins only after attaining an IND, RAC
review, and IBC and IRB approval (Mendell, 2004). Other countries also have a
number of committees that must approve gene therapy protocols and address other
scientific and ethical concerns asso-ciated with clinical trials (Kong, 2004).
Vectors for clinical trials must be produced according to strict Good
Laboratory Practice (GLP) and Good Manufacturing Practice (GMP) principles.
General requirements are described in 21 CFR and guidelines for the production
and qualification of cell and gene therapy products can be found in Points toConsider or
Guidance documents (www.fda.gov).
ICHguidance documents for quality-related issues also have application to gene
therapy products. Some of these documents are reproduced as chapters in the
United States Pharmacopeia (USP) (Seaver, 2000). Reference standards for gene
therapy vectors to assist manufacturers in characterizing raw materials,
pro-cess components and process impurities and for comparative analysis among
agents used in different clinical trials are needed. To date, an adenovirus
reference standard has been produced and is available through the American Type
Culture Collection (ATCC) (Hutchins, 2002). Reference standards for AAV and
lentivirus vectors are currently under development (Kiermer, 2005; Flotte,
2006).
Related Topics