Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Monoclonal Antibodies: From Structure to Therapeutic Application
Prediction of Human PK/PD Based on Preclinical Information
Prediction of Human PK/PD Based on Preclinical Information
Prior to first human studies, several preclinical in vivo and in vitro
experiments are performed to understand the PK of potential new drugs as well
as their safety and efficacy in animal models. However, the ultimate goal is at
all times to predict how these preclinical results on PK, safety and efficacy
translate into a given patient population. Therefore, predictions of human PK,
safety and efficacy are the focus of early drug development acknowledging the
similarities and differences between preclinical models and the respective
patient populations (see section “Preclinical Safety Assessment of mAbs”).
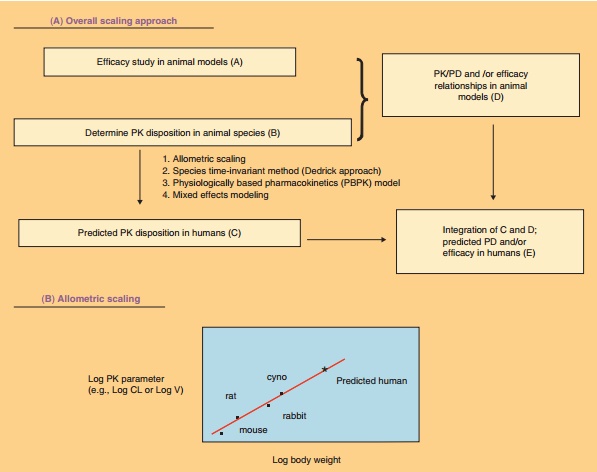
Over the years, many theories and different approaches have been
proposed and used for scaling preclinical data to clinical data. Figure 8
illustrates the prediction of human PK/PD based on preclinical information.
Allometric scaling is the simplest and most widely used method, which is based
on the power law relationship between body size and physiological and
anatomical parameters. This can be described by equation: Y¼a⋅ BWb, where Y
is the PK parameter (such as CL, V); BW is the
bodyweight; a is the coefficient; b is the exponent of the
allometric equations. Maximum life potential, brain weight, and two-term power
equation have been proposed as correction factors to improve the predic-tion
for CL. The accuracy of prediction of PK parameters by allometric scaling is
dependent on many factors, such as species, experimental design, analytical
errors, and others (Mahmood, 2005; Tangand Mayersohn, 2005). However, there are
only few reports on PK predictions using allometric scaling for mAbs. Lin et
al. (1999) projected the CL of bevacizu-mab in human as ~2.4 mL/day/kg based on
simple allometric scaling principles. Also Kelley et al. (2006) used simple
allometry to predict the CL of the anti-CD40 mAb of about 12 mL/day/kg in
humans. For these two examples, the human clearance was con-firmed to be in
agreement with the preclinical prediction by allometry.
Another approach for interspecies scaling is physiologically based
pharmacokinetic (PBPK) mod-eling, which establishes the animal PK based on
preclincial in vitro and in vivo data in the first step. In the second step,
the model is then scaled to humans by using human physiological information
such as blood flow, tissue volumes as well as potentially some additional human
in vitro data. Although PBPK modeling provides a mechanism based evaluation of
drug disposition, this approach is costly, mathemati-cally complex and time
consuming. Therefore in drug development and discovery, PBPK models are not as
widely used compared to allometry. However, for mAb interspecies scaling, PBPK
modeling allows to explore some physiological factors by simulation technology
which can not be incorporated into empirical allometric scaling methods, such
as the binding affinity, binding kinetics and non-linear PK. Baxter et al.
(1995) used PBPK models to predict PK of mAb against carcinoembryonic antigen
in human in different tissues including tumor compartments. It also has been
shown that a PBPK model including FcRn components worked very well to describe
the PK of an intact mAb and its Fab in different tissues in mice, with or
without tumor (Ferl et al., 2005).
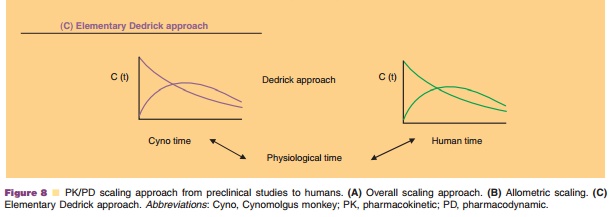
In addition, species-invariant time method (Mahmood, 2005) and
non-linear mixed-effect model-ing (Martin-Jimenez and Riviere, 2002; Jolling et
al., 2005) have been used for interspecies scaling for small molecules.
Species-invariant time method, also called Dedrick approach, was first
described in 1973 (Dedrick, 1973). Physiological time, the time required to
complete a species independent physiological event, can be obtained by
transformation of chron-ological time into a species invariant time (equivalent
time, kallynochrons, apolysichron, and dieneti-chrons). In the case that PK
follows allometric principles the transformation of chronological to
physiological time should provide superimposed concentration-time profiles for
all species. In his pioneering work Dedrick demonstrated this for methotrexate
as model compound (Dedrick, 1973).
Interspecies allometric scaling can also be performed using a population approaches (non-linear mixed effect modeling techniques). Allometric coeffi-cients and exponents as well as the variability from inter-animals, intra-animals and inter-species and random errors can be estimated in one single step by using this approach.
Due to the complexity of PD, any extrapolation to human requires more
complex considerations than for PK. Through PK/PD modeling and integration the
interspecies scaling of PK, the PD in human may bepredicted if the PK/PD
relationship is assumed to be the same between the animal models and humans.
For example, a PK/PD model was first developed to optimize the dosing regimen
of the mAb against EGF/r3 using tumor bearing nude mice as an animal model of
human disease (Duconge et al., 2004). This PK/PD model was then integrated with
allometric
scaling to calculate the dosage schedule required in a potential
clinical trial to achieve a certain effect (Duconge et al., 2004).
In summary, species differences in antigen density, antigen–antibody
binding, and antigen ki-netics, differences in FcRn binding between species,
the immunogenicity and other factors need to be considered during PK/PD scaling
of mAbs from animals to humans.
Related Topics