Chapter: Pathology: Liver Pathology
Jaundice
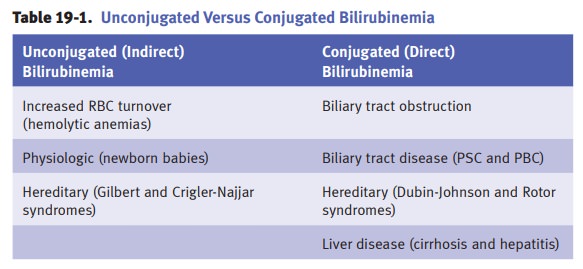
JAUNDICE
Clinical jaundice occurs with
bilirubin levels >2–3 mg/dL. The classic presentation is yellow skin
(jaundice) and sclera (icterus). Causes of jaundice include overpro-duction of
bilirubin, defective hepatic bilirubin uptake, defective conjugation, and
defective excretion.
Increased red blood cell (RBC) turnover. RBCs are the major source of bilirubin. Jaundice related to overproduction of bilirubin
can be seen in hemolytic anemia and ineffective erythropoiesis (thalassemia,
megaloblastic anemia, etc.). Laboratory studies show increased unconjugated
bilirubin. Chronic hemolytic anemia patients often develop pigmented
bilirubinate gallstones. The most common cause of marked jaundice in the
newborn is blood group incompatibility (most commonly ABO) between mother and
child, causing hemolytic disease of the
newborn.
Physiologic jaundice of the newborn is a transient unconjugated
hyperbilirubinemia due to the immaturity of the
liver. Risk factors include prematurity and hemolytic disease of the newborn
(erythroblastosis fetalis). Physiologic jaundice of the new-born can be
complicated by kernicterus; treatment is phototherapy. Jaundice also occurs in
newborns who have infections.
Hereditary hyperbilirubinemias
When hyperbilirubinemia is
prolonged in the newborn, a mutation affecting bili-rubin conjugation enters
the differential diagnosis.
•
Gilbert
syndrome is a common benign inherited disorder that causes uncon-jugated
hyperbilirubinemia due to bilirubin UDP-glucuronosyltransferase (UGT)
deficiency. Kernicterus rarely occurs and the treatment is phenobar-bital.
•
Crigler-Najjar
syndrome causes unconjugated hyperbilirubinemia due to bili-rubin
glucuronosyltransferase (UGT) absence or deficiency. Treatment for type 1 is
gene replacement therapy and liver transplantation. For a milder type 2,
phenobarbital is used.
•
Dubin-Johnson
syndrome is a benign autosomal recessive disorder
character-ized by decreased bilirubin excretion due to a defect in the
canalicular cationic transport protein. It produces conjugated
hyperbilirubinemia and a distinctive black pigmentation of the liver, but has
no clinical consequences.
•
Rotor
syndrome is an autosomal recessive conjugated hyperbilirubinemia that is similar to Dubin-Johnson syndrome, but
without liver pigmentation. There are no clinical consequences.
Biliary tract obstruction may have multiple etiologies,
including gallstones; tumors (pancreatic, gallbladder, and bile duct);
stricture; and parasites (liver flukes—Clo-norchis
[Opisthorchis] sinensis). The presentation can include jaundice and
icterus; pruritus due to increased
plasma levels of bile acids; abdominal pain, fever, and chills; dark urine
(bilirubinuria); and pale clay-colored stools. Lab studies show elevated
conjugated bilirubin, elevated alkaline phosphatase, and elevated
5´-nucleotidase.
Primary biliary cirrhosis (PBC) is a chronic liver
disease that is characterized by inflammation and
granulomatous destruction of intrahepatic bile ducts. Females have 10 times the
incidence of primary biliary cirrhosis compared to males; the peak incidence is
age 40–50.
Presentation includes
obstructive jaundice and pruritus; xanthomas, xanthelasmas, and elevated serum
cholesterol; fatigue; and cirrhosis (late complication). Most patients have
another autoimmune disease (scleroderma, rheumatoid arthritis or systemic lupus
erythematosis).
Laboratory studies show
elevated conjugated bilirubin, elevated alkaline phos-phatase, and elevated
5´-nucleotidase. Treatment with oral ursodeoxycholic acid slows disease
progression. Antimitochondrial
autoantibodies (AMA) are present in >90% of cases, which further supports
an autoimmune basis. Microscopically, lymphocytic and granulomatous
inflammation involves interlobular bile ducts.
Primary sclerosing cholangitis (PSC) is a chronic liver
disease characterized by segmental inflammation and
fibrosing destruction of intrahepatic and extrahepatic bile ducts. The exact
etiologic mechanism is not known but growing evidence sup-ports an immunologic
basis.
The male to female ratio is
2:1; peak age 20–40. Most cases of PSC are associated with ulcerative colitis.
The presentation is similar to PBC. Complications include biliary cirrhosis and
cholangiocarcinoma.
Microscopically, there is
periductal chronic inflammation with concentric fibro-sis around bile ducts and
segmental stenosis of bile ducts. Cholangiogram shows “beaded appearance” of
bile ducts.
Related Topics