Chapter: Modern Pharmacology with Clinical Applications: Histamine and Histamine Antagonists
Histamine Antagonism and Histamine Antagonists
HISTAMINE
ANTAGONISM AND HISTAMINE ANTAGONISTS
The effects of histamine on
body tissues and organs can be diminished in four ways: inhibition of histamine
syn-thesis, inhibition of histamine release from storage granules, blockade of
histamine receptors, and physio-logical antagonism of histamine’s effects. Of
these ap-proaches, only the inhibition of histamine synthesis has not been
employed clinically.
H1-Receptor Antagonists
The most common use of the H1-receptor
antagonists is for the relief of allergic reactions such as rhinitis and
ur-ticaria. These compounds are also used to prevent mo-tion sickness, to treat
vestibular disturbances, such as Ménière’s syndrome, and as over-the-counter
sleep aids.
Chemistry
The H1-receptor
antagonists for the most part are sub-stituted ethylamine compounds. In
comparison with his-tamine, the H1-antagonists contain no imidazole
ring and have substituents on the side chain amino group.
The H1-antagonists
are classified as either first- or second-generation compounds.
Second-generation anti-histamines have lipophilicity and ionization profiles
that make them less able to cross the blood-brain barrier; thus they produce
dramatically less sedation than do the first-generation drugs.
Pharmacokinetics
First-generation
antihistamines are well absorbed after oral administration, with peak blood
levels occurring within 1 to 2 hours; the therapeutic effect usually lasts 4 to
6 hours, although some drugs are much longer acting (Table 38.2). These
antagonists are generally metabo-lized in the liver through hydroxylation. The
metabo-lites and a small amount of parent compound are ex-creted in the urine.
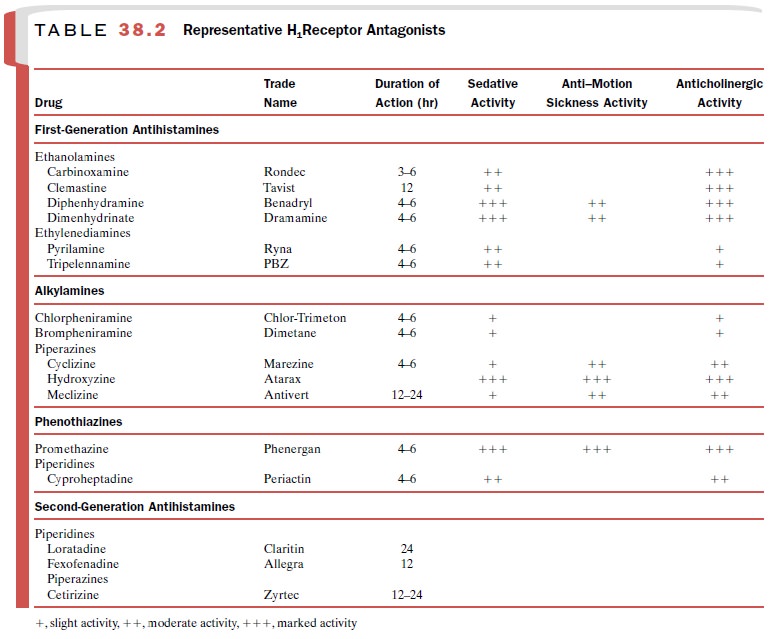
The second-generation H1-receptor
antagonists are also rapidly absorbed, with peak plasma concentrations being
reached within 1 to 3 hours. Their duration of ac-tion generally varies between
4 and 24 hours (Table 38.2). Loratadine (Claritin)
and its active metabolite, desloratadine (Clarinex),
undergoes extensive first-pass metabolism and is converted by CYP3A4 isozymes
to an active metabolite. A number of drug interactions re-sult from the ability
of various compounds to induce, in-hibit, or compete for metabolism by this
cytochrome P450 system. In contrast, cetirizine (Zyrtec) and fexofe-nadine (Allegra)
undergo little hepatic metabolism and are eliminated mainly as unchanged
compounds in the urine and feces, respectively.
The reduction in therapeutic
effectiveness that can occur when antihistamines are given for long periods is
probably related to an induction of hepatic drug-metabolizing enzymes. Children
tend to eliminate anti-histamines more rapidly than adults, while individuals
with hepatic impairment may eliminate them more slowly.
Mechanism of Action
At therapeutic doses, the first- and second-generation antihistamines are equilibrium-competitive inhibitors of H1-receptor–mediated responses. Certain second-generation drugs are noncompetitive inhibitors at high concentrations. Both first- and second-generation com-pounds have negligible abilities to block the H2-, H3-, or H4-receptors.
The therapeutic effectiveness of these drugs arises from their
capacity to block histamine-mediated vasoconstriction, microvascular permeability
enhancement, and sensory nerve terminal stimulation. H1-antagonists
generally produce sedation through an effect on the CNS; however, excitation
can occur when toxic dosages are ingested.
Many of these drugs have
effects that are not medi-ated by H1-receptors (Table 38.2). The
antimuscarinic ac-tivity of several first-generation H1-blockers may
account for their effectiveness in combating motion sickness and their limited
ability to suppress parkinsonian symptoms. The phenothiazines have some
capacity to block α- adrenoceptors, whereas cyproheptadine (Periactin) is an antagonist at serotonin receptors. Diphenhydramine
(Benadryl), pyrilamine (Ryna), and promethazine (Phen-ergan) are effective local
anesthetics. Many second-generation antihistamines also have been found to
in-hibit the non–histamine-mediated release of various inflammatory substances;
this may account for some of their effectiveness in allergic conditions.
Adverse Effects
Sedation is the most frequent
adverse reaction to the first-generation antihistamines. An additive effect on
alertness and motor skills will result if alcohol or an-other depressant is
taken with these drugs. Anti-muscarinic effects caused by these drugs include
dry mouth and respiratory passages, urinary retention, and dysuria. Nausea,
vomiting, constipation or diarrhea, dizziness, insomnia, nervousness, and
fatigue also have been reported. Drug allergy, especially after topical
ap-plication, is fairly common. Tolerance to certain antihis-tamines may develop
after prolonged administration. Teratogenic effects of the piperazine
antihistamines have been shown in animal studies. Epidemiologicalstudies have
not shown such an association in humans. The effects of toxic doses of
first-generatio n antihista-mines, similar to those seen following atropine
adminis-tration, include excitement, hallucinations, dry mouth, dilated pupils,
flushing, convulsions, urinary retention, sinus tachycardia, coma, and death.
The second-generation H1-antagonists
are often re-ferred to as nonsedating antihistamines; however, doses above the
usual therapeutic level can cause sleepiness in certain individuals. A more
serious adverse effect of some earlier second-generation antihistamines is
car-diotoxicity. Terfenadine (Seldane)
and astemizole (Hismanal) were
withdrawn from the U. S. market after they were found, in rare cases, to induce
a potentially fa-tal ventricular arrhythmia, torsades de pointes. These drugs block the cardiac K+ channels
responsible for the repolarizing current (IKr) of the action
potential and therefore prolong the QT
interval. Arrhythmias result when these drugs accumulate to toxic levels, such
as when their metabolism is impaired, as in liver disease or following
coadministration of drugs that inhibit the CYP3A family of enzymes.
Fexofenadine, the active antihistaminic metabolite of terfenadine, does not
produce torsades de pointes.
Clinical Uses
The H1-receptor
blocking drugs find their greatest use in the symptomatic treatment of allergic
conditions. The second-generation antihistamines and the first-generation
alkylamines are most frequently used to treat allergic rhinitis. Allergic
conjunctivitis and the acute form of urticaria are also effectively treated
with antihistamines. The allergic responses seen in suscepti-ble individuals
after intradermal injections of allergens (e.g., skin testing) can be prevented
for several hours by prior administration of H1-antagonists.
However, the H1-antagonists are not drugs of choice in acute
anaphy-lactic emergencies or the viral-caused common cold.
Although the antihistamines
are not useful as pri-mary agents in the treatment of asthma, a number of
studies have shown that the second-generation com-pounds are effective as
adjunctive therapies in asthmatic patients with concomitant rhinitis,
urticaria, or dermati-tis. Cetirizine has been used to prevent the progression
from atopic dermatitis to asthma in young children.
Another important use of H1-antagonists
is in the treatment of motion sickness. Diphenhydramine (Benadryl), dimenhydrinate (Dramamine),
cyclizine (Marezine), and meclizine (Antivert) have anticholiner-gic activity
and are the preferred antihistaminic agents for reducing the symptoms of motion
sickness.
Diphenhydramine is known to
be at least partially ef-fective in Parkinson’s disease, perhaps because of its
an-ticholinergic properties.
Many H1-receptor
blocking drugs have sedative properties, and some have been used in
over-the-counter sleep aids. The most widely used H1-blocking drugs
for sleep induction are diphenhydramine, pro-methazine, and pyrilamine.
H2-Receptor Antagonists
The H2-receptor
blockers include cimetidine, famoti-dine, and ranitidine. These drugs are used
to decrease gastric acid secretion in the treatment of peptic ulcer,
gastroesophageal reflux disorder, and hypersecretory conditions, such as
Zollinger-Ellison syndrome.
Cromolyn and Nedocromil
Although cromolyn sodium (Intal) and nedocromil sodium (Tilade) are widely known for their
ability to prevent the release of histamine and other inflamma-tory mediators
by mast cells during the early response to antigen challenge, these drugs have
a wide variety of inhibitory effects on many cell types, including eosinophils,
neutrophils, monocytes, and neurons. Cromolyn sodium and nedocromil sodium are
used as pulmonary inhalants in the treatment of asthma. Nasal (Nasalcrom) and ophthalmic (Opticrom) preparations of cromolyn
sodium can be used to reduce the symp-toms of allergic rhinitis and
conjunctivitis.
New Directions in Antihistamine Therapy
None of the selective
agonists and antagonists of H3-receptors are available for clinical
use. Antagonists of H3-mediated inhibition of neurotransmission may
have potential in the treatment of CNS disorders, since ani-mal studies have
found that these compounds may en-hance learning, ameliorate learning deficits,
and de-crease seizure activity. H3-receptor agonists have been shown
to inhibit gastric acid release and block certain inflammatory processes. In
cardiac ischemia, they can prevent the arrhythmia and cardiac damage that may
result from norepinephrine overflow and thus may be useful in the treatment of
myocardial infarction. Selective agonists and antagonists of H4-receptors
are not yet available.
Related Topics