Chapter: Basic Radiology : Brain and Its Coverings
Exercise: Congenital Anomalies
EXERCISE 12-1.
CONGENITAL ANOMALIES
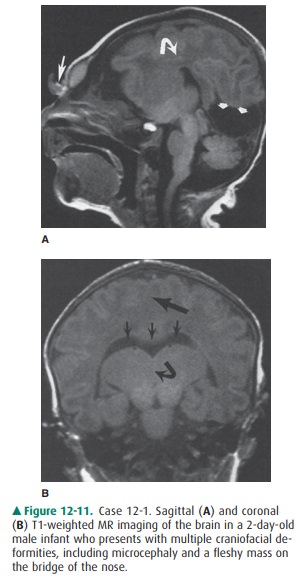
12-1. In Case 12-1, what is the major abnormality (Figure12-11
A, B)?
A.
Enlarged ventricles
B.
Cyst in the posterior fossa
C.
Lack of brain cleavage into two hemispheres
D.
Herniation of intracranial contents through a skull defect
E.
Abnormal migration of gray matter
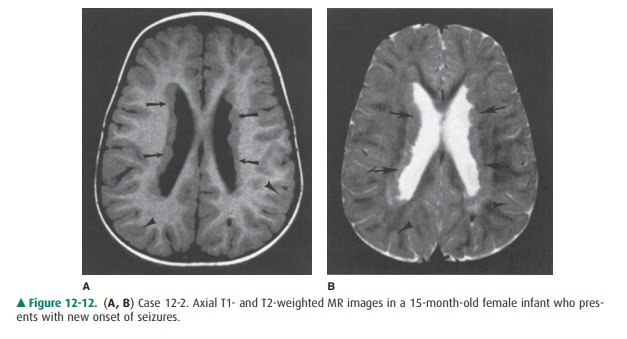
12-2. In Case 12-2, what is the etiology of the patient’s
seizures (Figure 12-12 A, B)?
A.
Brain tumor
B.
Gray matter in the wrong place (ie, heterotopic gray matter)
C.
Congenital infection
D.
Nodules along ventricles in a patient with tuber-ous sclerosis
E.
Infarction of periventricular white matter
Radiologic Findings
12-1. In this case, the corpus callosum (curved arrow) is absent on
the sagittal T1 MR image (Figure 12-11 A). Also note other midline
abnormalities, including ab-normal tissue at the bridge of the nose (large
arrow) and a posterior cyst (small arrows). Coronal T1-weighted MR image
(Figure 12-11 B) demonstrates a monoventricle (small arrows) and thalamic
fusion (curved arrow). Also note the lack of separation of the two hemispheres
(large arrow) (C is the correct answer to Question 12-1).
12-2. In this case, T1-weighted (Figure 12-12 A) and T2-weighted (Figure 12-12 B) MR images show abnor-mal tissue lining the lateral ventricle (arrows). Signal of this tissue follows that of normal gray matter (ar-rowheads) on both T1- and T2-weighted images (B is the correct answer to Question 12-2).
Discussion
Two common reasons for performing
MR scans in young in-fants are illustrated by the cases in this section.
Infants with craniofacial anomalies frequently have underlying congenital
malformations of the CNS. Seizures, too, may be the first sign of an underlying
brain malformation. As discussed in the section on technique selection,
whenever a congenital brain anomaly is suspected, MR imaging is the best examination
to perform.
Insults to the developing brain
lead to predictable alter-ations of brain morphology. By analyzing patterns of
al-tered brain morphology, we can often determine which stage of CNS
development has been disrupted. This analy-sis, combined with knowledge of
neuroembryology, has al-lowed for the development of systems to classify
congenital anomalies of the CNS. One simplified classification system divides
congenital malformations into disorders of organo-genesis (which include abnormalities
of neural tube closure, diverticulation/cleavage, sulcation/cellular migration,
and size, as well as destructive lesions acquired in utero), disor-ders of
histogenesis (ie, neurocutaneous syndromes), and disorders of cytogenesis (ie,
congenital neoplasms). Readers are referred to the suggested readings at the
end of this chap-ter for further information on this topic.
The patient in Case 12-1 has
alobar holoprosencephaly, a classic example of disordered ventral induction. In
this condi-tion, there is complete (alobar) or partial (semilobar, lobar)
fail-ure of separation of the forebrain (prosencephalon) into two hemispheres.
In alobar holoprosencephaly, the most severe form of this disorder, there is no
separation of the two hemi-spheres at all. The thalami are fused, a central
monoventricle is present, and there is no corpus callosum. Infants with this
form of holoprosencephaly frequently have severe facial anomalies.
In Case 12-2, the patient has
heterotopic gray matter lin-ing the lateral ventricles. This congenital anomaly
is one type of disordered cellular migration. Neurons that make up the gray
matter of the cerebral cortex actually develop along the edges of the lateral
and third ventricles within the so-called germinal matrix zone. They then migrate
outward to their final cortical location. If this normal neuronal migration is
disrupted, a normal cortex may not develop, and foci of gray matter may be
present in abnormal locations along the mi-gration route. Collections of these
normal neurons in abnor-mal locations are called gray matter heterotopias.
Several different types of
heterotopias have been described. The case presented in this section
demonstrates a focal nodu-lar gray matter heterotopia involving the
subependymal regionat the edge of the lateral ventricles. Seizures frequently
occur in patients with this condition, as in the patient in Case 12-2. Be-cause
MR imaging usually provides an exact diagnosis of this condition, biopsies of
CNS tissue are unnecessary.
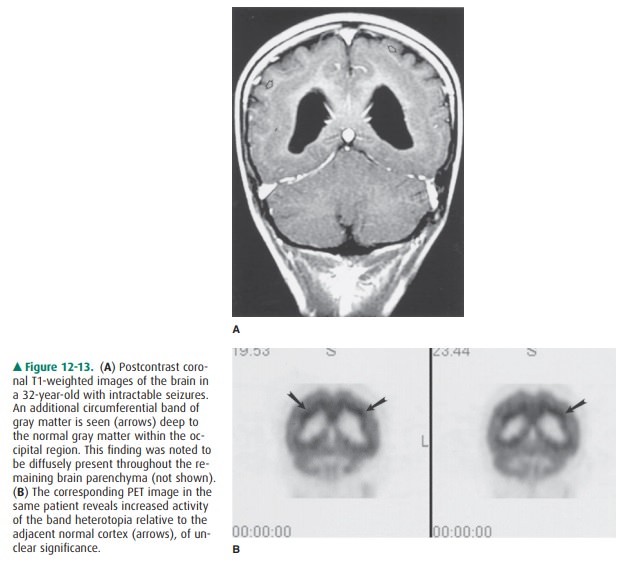
In contrast to focal nodular
heterotopias, diffuse (or laminar) heterotopias are commonly seen within or
adja-cent to the cortex, while “band” type heterotopias are lo-cated deep to
the normal cortex, in a subcortical location, separated by a thin interface of
white matter (Figure 12-13). Band-type heterotopias are well defined, with
smooth mar-gins, demonstrating signal intensities identical to those of normal
gray matter. Mass effect on the underlying white matter or deep gray structures
may be seen, and the sulca-tion pattern of the brain superficial to the
heterotopia may be abnormal. Associated CNS anomalies may be present, such as
agenesis of the corpus callosum, holoprosencephaly, or herniation of brain
tissue (encephaloceles). Although at first glance the cortex may appear to be
markedly thickened, closer examination will reveal an additional band of gray
matter in a subcortical location, which may or may not demonstrate increased 18F-FDG
activity on a PET scan. This band of heterotopia is known to be associated with
in-tractable seizures, occurring earlier than in the focal type, as well as
severe developmental delay.
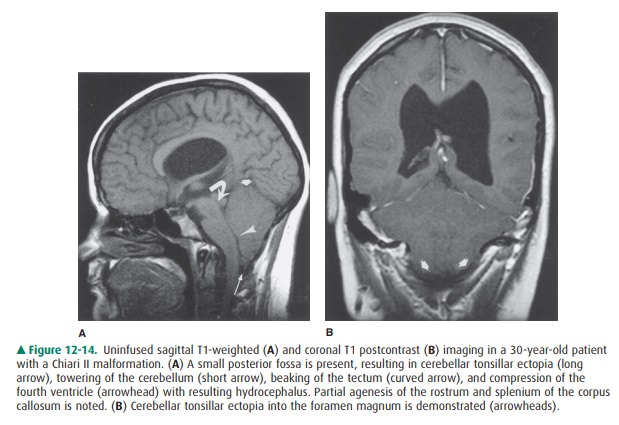
Several types of Chiari
malformations were initially de-scribed by the German pathologist Hans Chiari,
who clas-sified these congenital hindbrain anomalies into three types. In each
case, abnormal descent of cerebellar tissue into the cervical canal is
demonstrated. A Chiari I malfor-mation is associated with a relatively small
posterior fossa and a normal-sized cerebellum. Consequently, elongated peglike
cerebellar tonsils extend below the foramen mag-num with effacement of the
corresponding CSF spaces. There is often dorsal tilting of the dens, which may
indent the brainstem. There is no association between Chiari I malformations
and neural tube defects; however, the spine should be imaged because of the
common coexistence of a syrinx.
In contrast to Chiari I, the
Chiari II malformation is very highly associated with myelomeningocele and generally
supratentorial abnormalities. The posterior fossa is small with herniation of
cerebellar tonsils, vermis, and medulla below the foramen magnum. Because the
cervical cord is somewhat fixed in position by the dentate ligaments, this
downward displacement results in a characteristic cervi-comedullary kink. The
fourth ventricle is low-lying and elon- gated as well, with distortion of the
cerebral aqueduct and tectum (so-called tectal beaking), often resulting in
hydro-cephalus. The superior cerebellum towers superiorly through a widened
tentorium incisura, with the remainder of the cerebellum wrapping around the
brainstem. Supratentorial abnormalities include agenesis or hypoplasia of the
corpus callosum, enlarged massa intermedia, deficiency of the falx resulting in
interdigitation of cortical gyri across the midline, and enlarged occipital
horns (colpocephaly) (Figure 12-14). Chiari III malformations are associated
with occipital or high cervical encephaloceles, containing cerebellar tissue,
with or without brainstem.
Disorders of histogenesis include
the neurocutaneous syndromes, which are a heterogeneous group of disorders with
CNS and, for the most part, cutaneous manifesta-tions. Visceral and connective
tissue abnormalities may be prominent. Common disorders within this group
include neurofibromatosis types I and II, tuberous sclerosis, von Hippel-Lindau
disease, and Sturge-Weber syndrome, where the abnormal lesions corresponding to
these entities are neurogenic tumors, tubers, hemangioblastomas, and angiomas,
respectively.
Neurofibromatosis type 1 is the
most common of all the neurocutaneous syndromes, accounting for 90% of all
neu-rofibromatosis cases, and is the only entity discussed here. It is
transmitted on the long arm of chromosome 17 and is a disease of childhood.
Autosomal dominant transmission oc-curs in 50%, and the remainder appear
sporadically as new mutations in a patient with no known family history of the
disease. The diagnosis is established when two or more of the following
criteria are present: (1) six or more café-au-lait spots (brown skin
pigmentation), (2) two or more Lisch nod-ules (hamartomas) of the iris, (3) two
or more neurofibro-mas, (4) one or more plexiform neurofibromas, (5) axillary
freckling, (6) one or more bone dysplasias (ie, dysplasia of the greater
sphenoid wing), (7) optic nerve glioma, or (8) first-degree relative with
neurofibromatosis type 1.
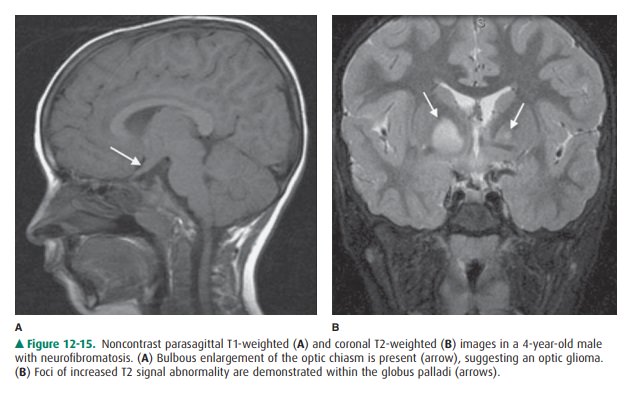
The optic pathway gliomas are
generally nonaggressive (low-grade) pilocytic astrocytomas, which present in
child-hood and may not affect vision until greatly increased in size (Figure
12-15 A). Cerebellar, brainstem, and cerebral astro-cytomas may additionally be
seen. High T2 signal intensity foci may be identified within the peduncles or
deep gray mat-ter of the cerebellum, brainstem, basal ganglia (particularly the
globus pallidus), and supratentorial white matter (Figure 12-15 B). The nature
of these lesions remains unresolved.
Related Topics