Chapter: Modern Pharmacology with Clinical Applications: Antihypertensive Drugs
Centrally Acting Hypotensive Drugs
CENTRALLY ACTING
HYPOTENSIVE DRUGS
Two important
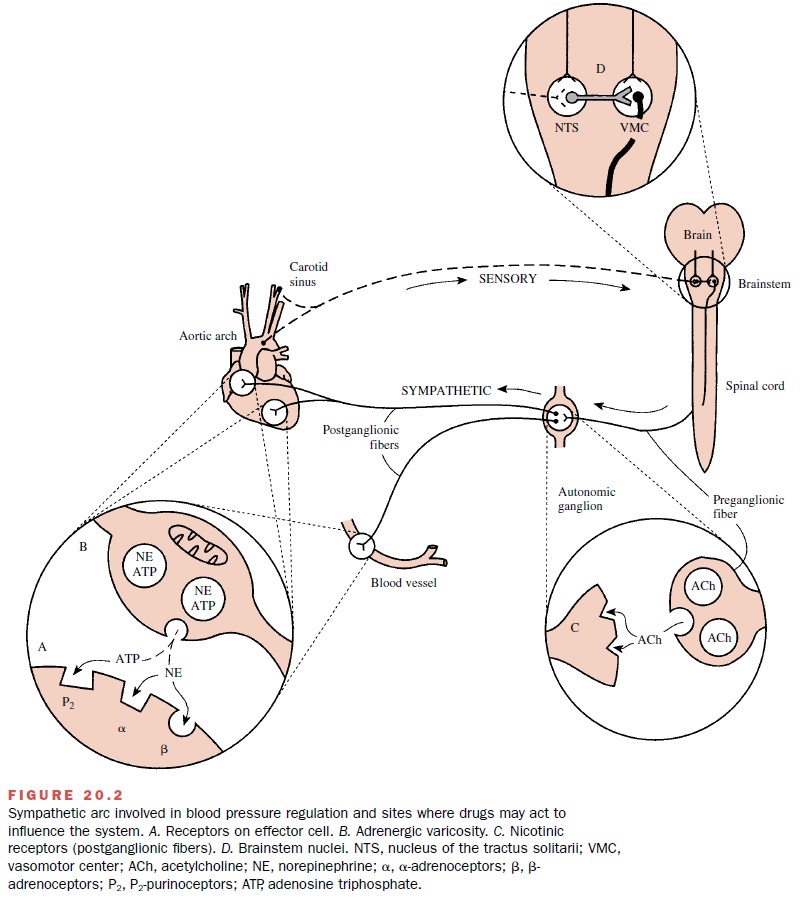
antihypertensive agents, α-methyldopa and clonidine, act predominantly in the brain (Fig. 20.2D). Although the details of their actions may differ in some
respects, their antihypertensive activity
is ulti-mately due to their ability to decrease the sympathetic outflow from
the brain to the cardiovascular system.
α-Methyldopa
The spectrum of activity of α-methyldopa
(Aldomet) lies between those of the
more potent agents, such as guanethidine, and the milder antihypertensives,
such as reserpine. α-methyldopa is a structural analogue of
di-hydroxyphenylalanine (dopa) and differs from dopa only by the presence of a
methyl group on the -carbon of the side chain.
Mechanism of Action
A number of theories have
been put forward to account for the hypotensive action of α-methyldopa. Current
evidence suggests that for α-methyldopa to be an anti-hypertensive agent, it
must be converted to -methyl-norepinephrine; however, its site of action appears to be in the brain rather than in the periphery. Systemically
ad-ministered α-methyldopa rapidly enters the brain, where it accumulates in
noradrenergic nerves, is con-verted to -methylnorepinephrine, and is released. Released -methylnorepinephrine activates CNS
α-adrenoceptors
whose function is to decrease sympathetic outflow. Why -methylnorepinephrine
decreases sym-pathetic outflow more effectively than does the natu-rally
occurring transmitter is not entirely clear.
Absorption, Metabolism, and Excretion
Approximately 50% of an
orally administered dose of α-methyldopa is absorbed from the gastrointestinal
tract. Both peak plasma drug levels and maximal blood pressure–lowering effects
are observed 2 to 6 hours af-ter oral administration. A considerable amount of
un-changed α-methyldopa and several conjugated and de-carboxylated metabolites
can be found in the urine.
Pharmacological Actions
The primary hemodynamic
alteration responsible for the hypotensive effects of α-methyldopa remains in
dis-pute. When the patient is supine, the reduction in blood pressure produced
by α-methyldopa correlates best with a decrease in peripheral vascular
resistance, car-diac output being only slightly reduced. When the pa-tient is
upright, the fall in blood pressure corresponds more closely with a reduced
cardiac output.
An important aspect of α-methyldopa’s
hemody-namic effects is that renal blood flow and glomerular fil-tration rate
are not reduced. As occurs with most sym-pathetic depressant drugs and
vasodilators, long-term therapy with α-methyldopa leads to fluid retention,
edema formation, and plasma volume expansion. While data conflict somewhat, it
is generally thought that α-methyldopa suppresses plasma renin activity.
Clinical Uses
α-Methyldopa is not generally
believed to be suitable for monotherapy of primary hypertension. Because plasma
volume increases as the duration of -methyl-dopa therapy is extended, the drug
should be used in conjunction with a diuretic; this will produce a
signifi-cantly greater fall in blood pressure than would occur with either drug
used alone. Because α-methyldopa low-ers blood pressure without compromising
either renal blood flow or the glomerular filtration rate, it is particu-larly valuable in hypertension complicated by renal
dis-ease. However, if end-stage renal failure accompanies se-vere
hypertension, α-methyldopa may not be effective.
The presence of α-methyldopa
and its metabolites in the urine reduces the diagnostic value of urinary
cat-echolamine measurements as an indicator of pheochro-mocytoma, since these
substances interfere with the flu-orescence assay for catecholamines.
Adverse Effects
The most commonly encountered
side effects of α-methyldopa are sedation and drowsiness. These CNS ef-fects
are probably the result of reductions in brain cat-echolamine levels. Other
side effects, also typical of sympathetic depression, are dry mouth, nasal
conges-tion, orthostatic hypertension, and impotence.
Autoimmune reactions
associated with -methyl-dopa treatment include thrombocytopenia and
leukope-nia. Since a few cases of an α-methyldopa–induced hep-atitis have
occurred, the drug is contraindicated in patients with active hepatic disease.
Flulike symptoms also are known to occur.
Clonidine and Related Drugs
Clonidine (Catapres) is effective orally and is
used pri-marily for the treatment of moderate hypertension. It is structurally
related to the α-adrenoceptor antagonists phentolamine and tolazoline. Clonidine, however, is not an α-blocker, but is actually an -agonist. Its
antihyper-tensive effectiveness appeared paradoxical until it was recognized
that clonidine activated central α 2-receptors, thus reducing sympathetic outflow
to the periphery.
Guanabenz (Wytensin) and guanfacine (Tenex) are two drugs with considerable
structural similarity to clonidine. These agents also are central α2-agonists and exhibit an
antihypertensive profile similar to that of clonidine.
Mechanism of Action
The antihypertensive activity of clonidine can be as-cribed solely
to a decrease in the sympathetic activity transmitted from the brain to the
peripheral vasculature. After clonidine administration, direct measurements of sympathetic
nerve activity show that electrical dis-charge is reduced in a number of
sympathetic nerves, in-cluding the cardiac, splanchnic, and cervical nerves.
It is generally agreed that
clonidine acts in the same general area in the brain as does α-methyldopa, that
is, somewhere in the medulla oblongata. The principal dif-ference between
clonidine and α-methyldopa is that clonidine acts directly on α2-receptors, whereas α-methyldopa first must be converted by
synthetic en-zymes to -methylnorepinephrine.
Absorption, Metabolism, and Excretion
Clonidine is well absorbed
after oral administration. Peak plasma levels occur between 2 and 4 hours after
drug administration and correlate well with pharmaco-logical activity. The
plasma half-life in patients with nor-mal renal function is 12 hours. Urinary excretion
of clonidine and its metabolites accounts for almost 90% of the administered
dose, and fecal excretion accounts for the rest. Approximately 50% of an
administered dose is excreted unchanged; the remainder is oxida-tively
metabolized in the liver.
Pharmacological Actions
An acute intravenous
injection of clonidine may pro-duce a transient pressor response that
apparently is due to stimulation of peripheral vascular α -receptors. The pressor
response does not occur after oral administra-tion, because the drug’s
centrally mediated depressor action overrides it.
The decrease in blood
pressure produced by cloni-dine correlates better with a decreased cardiac
output than with a reduction in peripheral vascular resistance. The reduction
in cardiac output is the result of both a decreased heart rate and reduced
stroke work; the lat-ter effect is probably caused by a diminished venous
return.
Renal blood flow and
glomerular filtration are not decreased, although renal resistance is
diminished. Like α-methyldopa, it is a useful agent for hypertension
complicated by renal disease. Plasma renin activity is re-duced by clonidine,
presumably as a result of a centrally mediated decrease in sympathetic
stimulation of the juxtaglomerular cells of the kidney.
Clinical Uses
The primary indication for
clonidine use is in mild and moderate hypertension that has not responded
ade-quately to treatment with a diuretic or a β-blocker. Since clonidine causes
sodium and water retention and plasma volume expansion, it generally is administered
in combi-nation with a diuretic. A vasodilator can be added to the
clonidine–diuretic regimen in the treatment of resistant forms of hypertension.
Such drug combinations can be quite effective, since the reflex increases in
heart rate and cardiac output that result from vasodilator adminis-tration are
reduced or negated by clonidine-induced de-creases in heart rate and cardiac
output.
For severely hypertensive
patients, clonidine has been used in combination with a diuretic, a
vasodilator, and a β-blocker. Some care must be taken, however, be-cause the
coadministration of clonidine and a β-blocker may cause excessive sedation.
Clonidine is especially useful in patients with renal failure, since its
duration of action is not appreciably altered by renal disease and it does not
compromise renal blood flow.
Adverse Effects
It is estimated that about 7%
of patients receiving cloni-dine discontinue the drug because of side effects.
Although the symptoms are generally mild and tend to subside if therapy is
continued for several weeks, as many as 50% of the patients complain of
drowsiness and dryness of mouth. Other untoward effects include constipation,
nausea or gastric upset, and impotence. These effects are characteristic of
interference with the functioning of the sympathetic nervous system.
A potentially dangerous
effect is rebound hyperten-sion, which
follows abrupt withdrawal of clonidine ther-apy. This posttreatment
hypertension appears to be the result of excessive sympathetic activity. The
genesis of the syndrome is not well understood. A contributing factor may be
development of supersensitivity in either the sympathetic nerves or the
effector organs of the car-diovascular system due to the clonidine-caused
chronic reduction in sympathetic activity. Thus, when the drug is abruptly
withdrawn, an exaggerated response to “nor-mal” levels of activity may occur.
If treatment with clonidine is terminated gradually, rebound hyperten-sion is
unlikely. Patients should be warned of the danger of abruptly discontinuing
clonidine treatment.
Related Topics