Chapter: Biotechnology Applying the Genetic Revolution: Aging and Apoptosis
AlzheimerŌĆÖs Disease
ALZHEIMERŌĆÖS
DISEASE
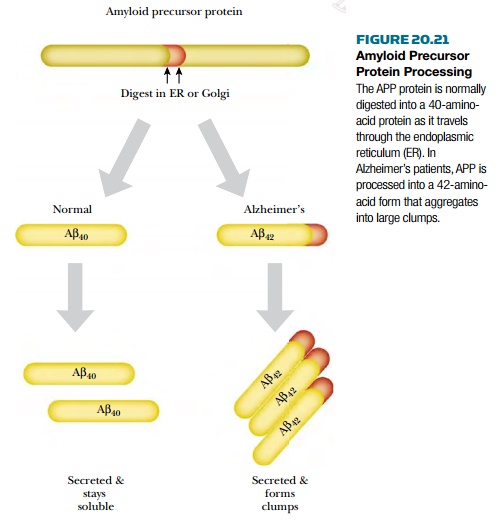
AlzheimerŌĆÖs disease is identified by two pathological hallmarks in the brain: senile neuritic plaques and neurofibrillary tangles. The senile neuritic plaques form when degenerating neurons aggregate into large clumps. The aggregates contain protein fragments known as amyloid ╬▓ ( A ╬▓ ) that are derived from amyloid precursor protein (APP). Normally, the precursor protein is digested by an enzyme in the endoplasmic reticulum to form a 40-amino-acid form called A ╬▓40 . In AlzheimerŌĆÖs, the precursor is digested in the wrong spot, and an aberrant 42-amino-acid form (A╬▓ 42 ) is made instead ( Fig. 20.21 ). This form tends to aggregate into clumps. In addition to the clumps of neurons, neurofibrillary tangles form inside otherwise normal neurons. This occurs when tau , a protein associated with microtubules, starts forming helical aggregates. These tangles eventually kill the neuron, which relies on the microtubules (and tau) to ship neurotransmitters from the cell body to the dendrites.
Beside tau and amyloid ╬▓ protein,
several other proteins are implicated in the pathology of AlzheimerŌĆÖs disease.
The presenilins are transmembrane proteins found in the endoplasmic reticulum
and Golgi apparatus. Mutations in presenilins may increase the secretion of the
A ╬▓42 form of amyloid ╬▓. Whether or not the
presenilins are the actual proteases that digest the amyloid precursor protein
remains to be proven. In addition, mutant presenilins make the nerve cell more
sensitive to apoptosis, although the mechanism is not understood. Mutations in
the presenilin genes, presenilin-1 and presenilin-2 , are the most
common defects in patients with familial AlzheimerŌĆÖs disease. These defects
also cause the disease to start earlier than usual. Another protein implicated
in AlzheimerŌĆÖs is apolipoprotein E. A specific allele of the gene for this ( e4) promotes the formation of plaques. If someone inherits
this allele, he or she is more likely to develop AlzheimerŌĆÖs after the age of
60.
The formation of senile neuritic plaques
causes the nerve cells to die, and the patient loses the function of that
region of the brain. When a plaque forms, the immune systemis alerted by
soluble signals from nearby microglial cells. These cells are activated by the aggregation
of amyloid ╬▓ protein. Once the immune system is activated, the immune
cells will kill more neurons by secreting proteins that activate the death
receptor pathway of apoptosis. (The immune system plays a role in the progress
of many neurodegenerative diseases.)
Because the role of amyloid ╬▓ in
plaque formation is a central aspect of AlzheimerŌĆÖs disease, recent work to
establish a treatment has focused on this molecule. Scientists have been able
to produce an antibody that breaks up aggregates of amyloid ╬▓ in mice.
The antibody can be given to AlzheimerŌĆÖs patients as a vaccine, and early
clinical trials have shown that it has few to no side effects. Although the
exact mechanism is unknown, clinical trials are under way to determine how
effective the vaccine is in human AlzheimerŌĆÖs patients. Potentially in 5 to 10
years a real treatment will exist for this devastating disease..
Related Topics